Pantox Laboratories, Inc.
4622 Santa Fe Street
San Diego, CA 92109
Client Services, Pantox Laboratories, Inc.
Copyright © 1998 Pantox Laboratories, Inc. All rights reserved.
|
Mark F. McCarty Pantox Laboratories, Inc. 4622 Santa Fe Street San Diego, CA 92109 |
Reprinted with the permission of Laura Ryan Client Services, Pantox Laboratories, Inc. Copyright © 1998 Pantox Laboratories, Inc. All rights reserved. |
This analysis reveals that both Turkel and Lawrence have administered high doses of folic acid as well as methyl donors (choline and betaine); the dose of pyridoxine employed is also relatively ample. Turkel recommended 5 mg folate daily; Lawrence has apparently used a high dose in the past, though her recommendation has recently been decreased to 800 µg. While Lawrence also places great emphasis on antioxidant nutrition, such is not the case with Turkel's formulation - thus, it is clear that antioxidants did not mediate the benefits claimed by Turkel. Since both folate and pyridoxine can have profound effects on mental function and brain development, it is intriguing to examine the published medical literature pertinent to folate and pyridoxine status in Down's syndrome.
With respect to pyridoxine, little of interest emerges in a literature search. However, there is uncontradicted evidence that the function of the monocarbon folate pool may be seriously impaired in Down's syndrome.1-3 The first clue in this regard came from the clinical observation that Down's patients were unusually sensitive to the toxic effects of the chemotherapeutic drug methotrexate, which depletes tetrahydrofolate by inhibiting dihydrofolate reductase. Subsequent in vitro studies by Lejeune and colleagues (celebrated for discovering that Down's is a gene overdosage syndrome) revealed that lymphocytes from Down's patients were roughly twice as sensitive to the mitosis-inhibiting action of methotrexate as lymphocytes from controls. Since tissue levels of folate per se are not notably decreased in Down's patients, and their enzymatic activity for thymidine monophosphate synthesis appears to be normal, it is reasonable to conclude that the monocarbon folate pool is deficient in these subjects. This is consistent with the observation that the erythrocytes of Down's patients are frequently found to be macrocytic4,5 - a characteristic symptom of folate deficiency. Lejeune and colleagues have demonstrated that high-dose folate supplementation in Down's patients can indeed substantially reduce the ex vivo sensitivity of their lymphocytes to methotrexate, presumably by a mass action effect.1
It is not yet clear why monocarbon folate status should be impaired in Down's patients. Lejeune has noted that three of the enzymes required for purine synthesis derive from chromosome 21;6-8 he postulates that this increases de novo purine synthesis in Down's patients, thereby boosting demand for monocarbons from the folate pool. There are in fact many reports that serum urate levels are elevated in Down's patients,9-12 and Lejeune's group has observed that plasma levels of serine (the chief source of carbon for the folate pool) are subnormal in these subjects.13 - observations that appear consistent with increased purine synthesis. However, it should be noted that the allosterically-regulated enzyme considered rate-limiting for de novo purine synthesis - amidophosphoribosyl transferase - is not coded on chromosome 21, so it is not straightforward to predict an increase in de novo purine synthesis in Down's. Furthermore, urate levels are not consistently elevated in Down's patients,14 and some investigators attribute the increase of serum urate in many Down's subjects to a decrease in renal clearance of urate.15 A report that adenosine deaminase activity is elevated in Down's patients.16 suggests that, if purine production is indeed increased in Down's, increased purine catabolism might be responsible, at least in part.
Since cystathionine beta-synthase (CBS) is coded on chromosome 21 and appears to be overexpressed in Down's, it has been suggested that a relative deficiency of homocysteine.17,18 may impede the demethylation of 5-methyltetrahydrofolate, such that a high proportion of the folate pool becomes trapped in this form and unavailable for other biosynthetic activities. Surprisingly, though, some investigators have found that homocysteine levels are normal in Downs, despite the overexpression of CBS.19 A possible explanation for this is that S-adenosylmethionine (an allosteric activator of CBS) is subnormal in Down's hepatocytes. If homocysteine levels are normal, or near-normal, in Down's syndrome, there is little reason to suspect that tetrahydrofolate is becoming "trapped" in its 5-methyl form - particularly inasmuch as B12 status appears to be normal in this disorder.
Although the biochemical basis for the dysfunction of the monocarbon folate pool in Down's remains murky, this does not detract from the potential pathogenic importance of this abnormality. Inherited metabolic abnormalities that impair folate function - as well as severe pre-natal and post-natal nutritional folate deficiency - typically are associated with mental retardation.20,21 It is notable that, in infants that have been severely folate deficient prenatally and postnatally, subsequent correction of folate status does not correct the mental retardation.21 Thus, impairment of folate function during critical periods of early development appears to have a permanent adverse impact on brain development that manifests as irreversible retardation.
Folate plays several key roles in the development and function of the CNS. Obviously, it is required for purine synthesis, as well as production of the thymidine required for cell multiplication and DNA repair. By maintaining adequate levels of S-adenosylmethionine, the folate pool promotes a range of methylation reactions, including those required for choline synthesis and catecholamine catabolism. A further activity of 5-methyltetrahydrofolate is to reduce dihydrobiopterin to tetrahydrobiopterin (a crucial catalyst of catecholamine synthesis) in a reaction catalyzed by methylenetetrahydrofolate reductase.22,23 It is thus readily understandable that effective folate function is critical for optimal development and function of the brain.
On the presumption that the monocarbon folate pool is indeed dysfunctional in Down's patients, Lejeune and colleagues have in fact administered very high does of folic acid (or folinic acid) - up to 1 mg/kg daily - to many such patients. They report that, in patents displaying psychotic behavior or Alzheimer's-type dementia, that such supplementation frequently has a dose-dependent favorable impact on behavior (3). However, unlike Turkel or Lawrence, they do not address the possibility that such supplementation, initiated as soon as possible after birth, might have a favorable impact on the subsequent physical and mental development of the general population of Down's infants.
In light of the fact that deficiency or dysfunction of the folate pool can lead to permanent brain damage manifesting as mental retardation, as well as compelling evidence of suboptimal folate function in Down's patients that can be compensated with high-dose supplemental folate, it would seem prudent to provide Down's infants with high-dose folate from the earliest age - in utero, if possible. It appears that the folate dose recommended by Lawrence has rather recently been reduced to 800 µg - on legal grounds, and on the basis of speculation that excess folate may impede dihydrobiopterin reduction. I am unable to find any evidence supporting this latter concern - indeed, 5-methyltetrahydrofolate can promote such reduction. It may therefore be unwise to use less than the 5 mg dose in Turkel's formulation (which in turn was often less than the dose used by Lejeune). 800 µg would be a fine dose if the intent were to correct folate deficiency - but in fact folate levels are usually normal in Down's syndrome; the rational intent of folate supplementation in Down's patients is to compensate for defective function of the monocarbon folate pool by providing tissues with supranormal amounts of folate.
Furthermore, in light of the role of serine as the chief carbon source for the monocarbon folate pool, and the report that serum folate levels are subnormal in Down's patients, it might be reasonable to provide supplemental serine in multi-gram daily doses (see diagram: Folate Road Map)
Biosynthesis of creatine in the liver and kidney consumes the large majority of the methyl groups used in biosynthetic reactions.24,25 In rats, exogenous creatine can down-regulate endogenous creatine synthesis, thus alleviating a very significant drain on the monocarbon folate pool.26,27 This suggests the possibility that adequate creatine supplementation might have a favorable impact on the function of this pool in Down's patients.
The most outstanding feature of the supplementation regimen currently applied by Lawrence is its antioxidant nutrition. The notion that Down's patients may be under increased antioxidant stress is supported by a report documenting increased urinary excretion of peroxidation by-products as well as of 8-hydroxyguanosine (indicative of increased radical damage to DNA).28 No less germane is a recent report that cortical neurons from Down's fetuses are unusually prone to apoptosis when cultured in vitro; this apoptosis is associated with increased free radical activity, and can be inhibited by a variety of free radical scavengers.29 Thus, there are reasonable grounds to suspect that improved antioxidant status might have a favorable impact on cerebral development in Down's syndrome. However, it is clear that the benefits claimed by Turkel are not mediated by antioxidants, as his supplementation program was mediocre in this respect.
In regard to oxidant stress in Down's syndrome, I cannot concur with the common presumption that a 50% overdosage of superoxide dismutase (SOD) is responsible for this stress. Pharmacologically, exogenous SOD is useful as an antioxidant measure in ischemia-reperfusion damage. In steady state, the rate at which a tissue generates hydrogen peroxide will be determined chiefly by the rate at which it evolves superoxide. If this superoxide is not dismutated enzymatically, it will dismutate "spontaneously" - with co-generation of dangerous singlet oxygen, and in the context of much higher equilibrium levels of superoxide. (Ironically, Turkel attributed the low incidence of atheroma in Down's patients to the antioxidant benefit of increased SOD - a view that has some credibility.) Although one research group has repeatedly published reports that over-expression of SOD in cells or in rodents exerts pathogenic effects associated with radical damage,30,31 other groups have reported protective antioxidant effects for such overexpression.32 A further consideration is that certain partially trisomic individuals expressing a Down's phenotype do not over-express SOD.33 If in fact Down's patients are under increased oxidant stress - as suggested by some data - this is probably attributable either to an unexplained increase in superoxide generation, or an unknown dysfunction of antioxidant defenses. In any case, the fact that we cannot adequately explain the origin of oxidant stress in Down's syndrome does not lessen the desirability of controlling such stress with antioxidant nutrition.
With regard to antioxidant stress in the CNS, there is recent evidence that coenzyme Q10 (CoQ) may be particularly beneficial as a brain antioxidant.34,35 Indeed, high-dose CoQ is now being tested as a therapy for Parkinson's disease, in which oxidant stress is clearly a major pathogenic factor. The regimen currently recommended by Lawrence provides 30 mg CoQ daily. CoQ supplements with greatly improved bioavailability - such as "Q-Gel" - have recently become commercially available,.36,37 and the use of such formulations in Down's infants might be appropriate. The fact that most commercial preparations of CoQ have limited bioavailability emphasizes the desirablility of directly assessing antioxidant status with serum assays such as those provided by Pantox Labs.
Whether or not high-dose folate, methyl donors, or antioxidants do indeed have a favorable impact on the mental and physical development of Down's children, the thesis that something is likely to prove helpful in this regard is intriguing and credible. Many inherited metabolic disorders reflect structural errors in key enzymes or regulatory factors that are not susceptible to correction or compensation. In contrast, the proteins synthesized by Down's patients are not structurally defective - the syndrome presumably reflects an imbalance in the level of expression of various proteins. It is plausible that certain nutritional or pharmaceutical measures could have a compensatory beneficial impact on the metabolic imbalances that mediate the dysfunctions in Down's syndrome, and that such measures, if initiated as soon as feasible (preferably in utero!) could promote normal mental and physical development. Thus, the quest undertaken by Turkel and Lawrence is both credible and commendable, and deserves far better than the dismissive attitude with which many "experts" have greeted it.
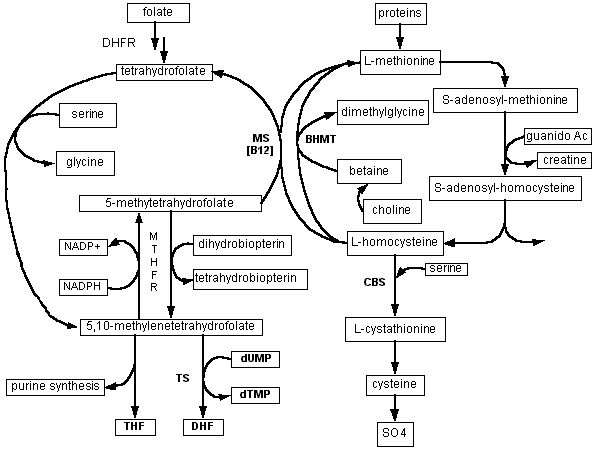
This chart depicts the metabolic roles of the monocarbon folate pool, and its relationship to methionine metabolism. 5,10-methylenetetrahydrofolate - produced when serine donates a carbon to tetrahydrofolate - provides carbon groups for synthesis of purines as well as deoxythymidine monophosphate (dTMP) - crucial for DNA synthesis and repair. (10-formyl tetrahydrofolate also participates in purine synthesis - not depicted here.)
5-methyltetrahydrofolate is the methyl donor for one of the two pathways (methionine synthetase = MS) in which homocysteine can be reconverted to methionine; this reaction also requires vitamin B12 as a cofactor. In the liver and kidney, betaine can serve as an alternative methyl donor for resynthesis of methionine by the enzyme called betaine-homocysteine methyl transferase (BHMT). Methionine in turn, after conversion to S-adenosylmethionine, serves as methyl donor for a vast array of biosynthetic reactions, including DNA methylations and synthesis of creatine, phosphatidylcholine, and other crucial compounds. An additional role for 5-methyltetrahydrofolate is reduction of dihydrobiopterin to tetrahydrobiopterin; the latter is required for the hydroxylation reactions involved in neurotransmitter synthesis (catecholamines and serotonin).
Why the efficiency of the monocarbon folate pool is impaired in Down's syndrome is not yet clear. Possible explanations include an excessive rate of purine synthesis, which draws carbons from the pool, as well as a relative deficiency of homocysteine (secondary to overexpression of the enzyme cystathionine-beta-synthase), which may result in "trapping" of folate in the 5-methyltetrahydrofolate form.
References
1. Peeters MA, Rethore MO, Lejeune J. In vivo folic acid supplementation partially corrects in vitro methotrexate toxicity in patients with Down syndrome. Br J Haematol 1995;89:678-680.
2. Lejeune J, Peeters M, Rethore MO, de Blois MC. Homocysteine and the methotrexate toxicity in trisomy 21 [letter]. Cancer Chemother Pharmacol 1991;27:331-332.
3. Lejeune J, Rethore MO, de Blois MC, Peeters M. [Infantile psychosis, pseudo-Alzheimer syndrome, and trisomy 21. A trial of treatment with folinic acid: preliminary report]. Therapie 1989;44:115-121.
4. Roizen NJ, Amarose AP. Hematologic abnormalities in children with Down syndrome. Am J Med Genet 1993;46:510-512.
5. David O, Fiorucci GC, Tosi MT, et al. Hematological studies in children with Down syndrome. Pediatr Hematol Oncol 1996;13:271-275.
6. Patterson D, Graw S, Jones C. Demonstration, by somatic cell genetics, of coordinate regulation of genes for two enzymes of purine synthesis assigned to human chromosome 21. Proc Natl Acad Sci U S A 1981;78:405-409.
7. Hards RG, Benkovic SJ, Van Keuren ML, Graw SL, Drabkin HA, Patterson D. Assignment of a third purine biosynthetic gene (glycinamide ribonucleotide transformylase) to human chromosome 21. Am J Hum Genet 1986;39:179-185.
8. Brodsky G, Barnes T, Bleskan J, Becker L, Cox M, Patterson D. The human GARS-AIRS-GART gene encodes two proteins which are differentially expressed during human brain development and temporally overexpressed in cerebellum of individuals with Down syndrome. Hum Mol Genet 1997;6:2043-2050.
9. Kaufman JM, O'Brien WM. Hyperuricemia in mongolism. N Engl J Med 1967;276:953-956.
10. Pant SS, Moser HW, Krane SM. Hyperuricemia in Down's syndrome. J Clin Endocrinol Metab 1968;28:472-478.
11. Appleton MD, Haab W, Burti U, Orsulak PJ. Plasma urate levels in mongolism. Am J Ment Defic 1969;74:196-199.
12. Nishida Y, Akaoka I, Nishizawa T, et al. A case of gouty arthritis associated with Down's syndrome. J Ment Defic Res 1976;20:277-283.
13. Mircher C, Salabelle A, Peeters MA, et al. [Variation of amino acids in relation to age in Down syndrome subjects]. Arch Pediatr 1997;4:1093-1099.
14. Watts RW, Perera YS, Allsop J, Newton C, Platts-Mills TA, Webster AD. Immunological and purine enzyme studies on hyperuricaemic and normouricaemic patients with Down's syndrome. Clin Exp Immunol 1979;36:355-363.
15. Nishida Y, Akaoka I, Kobayashi M, Maruki K, Oshima Y. Renal impairment in urate excretion in patients with Down's syndrome. J Rheumatol 1979;6:103-107.
16. Puukka R, Puukka M, Leppilampi M, Linna SL, Kouvalainen K. Erythrocyte adenosine deaminase, purine nucleoside phosphorylase and phosphoribosyltransferase activity in patients with Down's syndrome. Clin Chim Acta 1982;126:275-281.
17. Brattstrom L, Englund E, Brun A. Does Down syndrome support homocysteine theory of arteriosclerosis? [letter]. Lancet 1987;1:391-392.
18. Chadefaux B, Ceballos I, Hamet M, et al. Is absence of atheroma in Down syndrome due to decreased homocysteine levels? [letter]. Lancet 1988;2:741.
19. Brattstrom L, Israelsson B, Tengborn L, Hultberg B. Homocysteine, factor VII and antithrombin III in subjects with different gene dosage for cystathionine beta-synthase. J Inherit Metab Dis 1989;12:475-482.
20. Erbe RW. Inborn errors of folate metabolism (second of two parts). N Engl J Med 1975;293:807-812.
21. Greenblatt JM, Huffman LC, Reiss AL. Folic acid in neurodevelopment and child psychiatry. Prog Neuropsychopharmacol Biol Psychiatry 1994;18:647-660.
22. Coppen A, Swade C, Jones SA, Armstrong RA, Blair JA, Leeming RJ. Depression and tetrahydrobiopterin: the folate connection. J Affect Disord 1989;16:103-107.
23. Bottiglieri T, Hyland K, Laundy M, et al. Folate deficiency, biopterin and monoamine metabolism in depression. Psychol Med 1992;22:871-876.
24. Mudd SH, Poole JR. Labile methyl balances for normal humans on various dietary regimens. Metabolism 1975;24:721-735.
25. Mudd SH. Diseases of sulphur metabolism: implications for the methionine- homocysteine cycle, and vitamin responsiveness. Ciba Found Symp 1979;239-258.
26. Balsom PD, Soderlund K, Ekblom B. Creatine in humans with special reference to creatine supplementation. Sports Med 1994;18:268-280.
27. McCarty MF. Supplemental creatine may decrease serum homocysteine and abolish the homocysteine "gender gap" by suppressing endogenous creatine synthesis. Submitted for publication 1999;
28. Jovanovic SV, Clements D, MacLeod K. Biomarkers of oxidative stress are significantly elevated in Down syndrome. Free Radic Biol Med 1998;25:1044-1048.
29. Busciglio J, Yankner BA. Apoptosis and increased generation of reactive oxygen species in Down's syndrome neurons in vitro. Nature 1995;378:776-779.
30. Peled-Kamar M, Lotem J, Okon E, Sachs L, Groner Y. Thymic abnormalities and enhanced apoptosis of thymocytes and bone marrow cells in transgenic mice overexpressing Cu/Zn-superoxide dismutase: implications for Down syndrome. EMBO J 1995;14:4985-4993.
31. Groner Y, Elroy-Stein O, Avraham KB, et al. Cell damage by excess CuZnSOD and Down's syndrome. Biomed Pharmacother 1994;48:231-240.
32. Schwartz PJ, Coyle JT. Effects of overexpression of the cytoplasmic copper-zinc superoxide dismutase on the survival of neurons in vitro. Synapse 1998;29:206-212.
33. De La Torre R, Casado A, Lopez-Fernandez E, Carrascosa D, Ramirez V, Saez J. Overexpression of copper-zinc superoxide dismutase in trisomy 21. Experientia 1996;52:871-873.
34. Matthews RT, Yang L, Browne S, Baik M, Beal MF. Coenzyme Q10 administration increases brain mitochondrial concentrations and exerts neuroprotective effects. Proc Natl Acad Sci U S A 1998;95:8892-8897.
35. Beal MF, Matthews RT, Tieleman A, Shults CW. Coenzyme Q10 attenuates the 1-methyl-4-phenyl-1,2,3,tetrahydropyridine (MPTP) induced loss of striatal dopamine and dopaminergic axons in aged mice. Brain Res 1998;783:109-114.
36. Singh RB, Wander GS, Rastogi A, et al. Randomized, double-blind placebo-controlled trial of coenzyme Q10 in patients with acute myocardial infarction. Cardiovasc Drugs Ther 1998;12:347-353.
37. Chopra RK, Goldman R, Sinatra ST, Bhagavan HN. Relative bioavailability of coenzyme Q10 formulations in human subjects. Int J Vitam Nutr Res 1998;68:109-113.
| Revised: March 21, 2000. |