Kjerstin M. Owens
Free Radical and Radiation Biology
The University of Iowa
Iowa City, IA 52242-1181
May 5, 2005
|
 |
Reproduced with the permission of the author
Free Radicals in Biology and Medicine (77:222, Spring 2005)
Free Radical and Radiation Biology Program
B-180 Med Labs
Instructors: Garry R. Buettner, Ph.D. Larry W. Oberley, Ph.D. with guest lectures from: Drs. Freya Q . Schafer, Douglas R. Spitz, and Frederick E. Domann
|
The Fine Print:
Because this is a paper written by a beginning student as an assignment, there are no guarantees that everything is absolutely correct and accurate.
In view of the possibility of human error or changes in our knowledge due to continued research, neither the author nor The University of Iowa nor any other party who has been involved in the preparation or publication of this work warrants that the information contained herein is in every respect accurate or complete, and they are not responsible for any errors or omissions or for the results obtained from the use of such information. Readers are encouraged to confirm the information contained herein with other sources.
All material contained in this paper is copyright of the author, or the owner of the source that the material was taken from. This work is not intended as a threat to the ownership of said copyrights.
Abbreviations
AMKL — Acute megakaryoblastic leukemia MnSOD — Manganese superoxide dismutase
AML — Acute myeloid leukemia mtDNA — Mitochondrial DNA
ALL — Acute lymphoblastic leukemia PET — Positron emission tomography
DS — Down's Syndrome RNAi —RNA interference
CuZnSOD — Copper zinc superoxide dismutase ROS — Reactive oxygen species
GPx — Glutathione peroxidase SOD — Superoxide dismutase
HIF-1 — Hypoxia inducible factor-1 VEGF — Vascular endothelial growth factor
MnSOD — Manganese superoxide dismutase
mtDNA — Mitochondrial DNA
PET — Positron emission tomography
RNAi —RNA interference
ROS — Reactive oxygen species
SOD — Superoxide dismutase
VEGF — Vascular endothelial growth factor
Table of Contents
- Introduction of Down's syndrome
- Incidence of cancer
- Increase in leukemia
- Decrease in solid tumors
- Trisomy 21
- CuZnSOD
- Type XVIII collagen and endostatin
- Tiam1
- GATA1
- Experimental hypotheses
- In vitro transformation of Down's syndrome cells
- In vivo test of solid tumor formation
- Conclusions
- References
Abstract
Down's syndrome is a genetic disorder that is caused by trisomy of chromosome 21. Individuals with Down's syndrome have an increase risk of leukemia (10-20 fold) and a decreased incidence of solid tumors. It has been speculated that the genes on chromosome 21 are responsible for this abnormal distribution of cancer. One of such genes is copper zinc superoxide dismutase which catalyzes the dismutation of superoxide into hydrogen peroxide. Endostatin, a potent angiogenesis inhibitor, is also upregulated by trisomy 21. The paper discusses the possible role of these two genes in the occurrence of cancer in Down's syndrome subjects.
Introduction of Down's Syndrome
Down's syndrome (DS) is the most common genetic disorder with an incidence of 1 in 700 births [1]. It is characterized by the physical features of short stature, hypotonia in the neonate, epicanthic eye folds, and a protruding tongue [2]. Cognitive impairment along with mental retardation are the defining characteristics of the disorder [1,2]. People with DS age quicker and typically do not live past their thirties. Those who do live past thirty have a severe increase in Alzheimer's dieseas [2]. These people also experience a myriad of medical complications, such as gastrointestinal malformations, congenital heart disease, and leukemia [1,3].
Down's syndrome was first described in 1866 [3]. It was in 1959 that the disorder was first linked to trisomy 21 [3]. Cells that have trisomy 21 have a third copy of chromosome 21. The genes found on this chromosome have been hypothesized to cause Down's syndrome and the complications of the disorder. Prenatal screening tests are available to determine the karyotype of the child in utero, however, these tests are typically only administered to high-risk pregnancies.
This paper will discuss the increase incidence of leukemia and decreased occurrence of solid tumors in DS subjects, and look at some of the genes postulated to be involved. We will discuss the implications of the upregulation of a handful of genes found on chromosome 21q22, including two genes that are often associated with cancer, copper zinc superoxide dismutase and endostatin, and two genes that act like oncogenes in lymphatic cells, Tiam1 and GATA1. Experimental hypotheses will also be proposed to examine whether reactive oxygen species (ROS) are involved in the occurrence of cancer in these individuals.
Incidence of cancer
Increase in leukemia
Children with Down's syndrome have a 10 to 20 fold increased risk of leukemia over non-DS children [1,3-5]. Moreover, they have a 500-fold increased risk of acute megakaryoblastic leukemia (AMKL) [1,3]. AMKL is a type of acute myeloid leukemia (AML) that arises from megakaryocyte precursor cells [3]. This specific type of leukemia makes up 6% of all AML cases in non-DS patients, whereas, is DS patients it accounts for 62% of AML cases [3]. Hasle and colleagues looked at the incidence of cancer in 2814 Down's syndrome patients in the Danish Cytogenic Registry cross-referenced with the Danish Cancer Registry (Table 1). They found a large increase in all leukemia, standardized incidence ratio of 17.63 [4].
Down's syndrome patients not only have an increase in leukemia, they also have an earlier age of onset. In fact, leukemia has been known to arise in a DS neonate [5]. Several newborns with DS also develop what is known as transient leukemia. This may then develop into AMKL [1].
Decrease in solid tumors
Hasle found that the incidence of solid tumors in DS patients was decreased as compared to the expected rate (Table 1). However, there was an increase in solid tumors in the peritoneum, ovaries, bladder, testis, and eyes [4]. The increase in ovarian cancer is consistent with reports of increased germ cell cancers in patients with DS [6]. Additionally, Satge et al. looked at the incidence of neuroblastoma in 6724 DS children. They did not find a single case of neuroblastoma in all of the subjects, although the expected frequency of the tumor was more than five [7].
This decrease in solid tumors in DS subjects has been of great interest to the scientific community, particularly in cancer research. However, this lack of solid tumor occurrence is very contradictory to the incidence in leukemia in these patients.
Table 1. Incidence of cancers in 2814 Down's syndrome patients in Denmark. Observed and expected values are used to calculate the standardized incidence ratio [4]
| Site (ICD-7) |
Observed |
Expected |
SIR (95% CI) |
| All sites (140-205) |
60 |
49·83 |
1·20 (0·92-1·55) |
| Buccal cavity (140-148) |
0 |
1·04 |
0·00 (0·0-2·88) |
| Digestive system (150-159) |
4 |
6·52 |
0·61 (0·17-1·57) |
| Stomach (151) |
1 |
0·91 |
1·10 (0·01-6·14) |
| Colon (153) |
2 |
2·24 |
0·89 (0·10-3·23) |
| Peritoneum (158-159) |
1 |
0·01 |
67·77 (0·89-3·77) |
| Respiratory system (160-164) |
1 |
4·96 |
0·20 (0-1·12) |
| Lung (162) |
1 |
4·96 |
0·20 (0-1·12) |
| Breast (170) |
0 |
7·32 |
0·00 (0·0-0·41) |
| Female genital organs(177-179) |
4 |
5·68 |
0·70 (0·19-1·80) |
| Uterus (172) |
1 |
1·20 |
0·83 (0·01-4·63) |
| Ovary (175) |
3 |
1·52 |
1·97 (0·40-5·5·77) |
| Male genital organs (177-179) |
4 |
2·82 |
1·42 (0·38-3·63) |
| Testis (178) |
4 |
2·15 |
1·86 (0·50-4·77) |
| Urinary tract (180-181) |
4 |
2·97 |
1·35 (0·36·-3·45) |
| Kidney (180) |
1 |
1·19 |
0·84 (0·01-4·66) |
| Bladder (181) |
3 |
1·78 |
1·69 (0·34-4·93) |
| Skin (190-191) |
2 |
8·14 |
0·25 (0·03-0·89) |
| Non-melanoma |
2 |
5·76 |
0·35 (0·04-1·25) |
| Other specified sites (192-197) |
2 |
4·75 |
0·42 (0·05-1·52) |
| Eye (192) |
1 |
0·27 |
3·68 (0·05-20·5) |
| Brain (193) |
1 |
0·27 |
3·32 (0-1·68) |
| Secondary and unspecified sites (198-199) |
3 |
0·92 |
3·27 (0·66-9·56) |
| Non-Hodgkin lymphoma (200, 202) |
0 |
1·41 |
0·00 (0·0-2·13) |
| Hodgkin's disease (201) |
0 |
0·92 |
0·00 (0·0-3·25) |
| All solid tumours (140-202) |
24 |
47·77 |
0·50 (0·32-0·75) |
| Leukaemia (204) |
36 |
2·04 |
17·63 (12·4-24·4) |
| ALL |
20 |
0·82 |
24·36 (14·9-37·6) |
| AML |
12 |
0·59 |
20·28 (10·5-35·4) |
| Acute unspecified leukaemia |
3 |
0·11 |
26·86 (5·4-78·5) |
| Leukaemia not otherwise specified |
1 |
0·52 |
1·93 (0·03-10·8) |
Trisomy 21
It has long been known that Down's syndrome is cause by three copies of chromosome 21. As much as 95% of DS cases occur through nondisjunction during meiosis which leads to a third copy of chromosome 21 [3]. This can occur either during meiosis I or II. Meiosis is a two cycle process similar to mitosis to form haploid gametes. Nondisjunction can occur in meiosis I if the chromosome pairs fail to separate during anaphase I. In meiosis II the sister chromatids fail to separate during anaphase II. Nondisjuction in either stage will lead to a gamete that is diploid for chromosome 21 and haploid for all of the other chromosomes. It has been shown that maternal nondisjunction (the ovary is the gamete with the extra chromosome) is more common and increases as a function of maternal age [3].
The other 5% of DS cases occur as a result of a chromosomal translocation that leads to an extra copy of chromosome 21q22 [3]. Chromosomal translocation occurs when two or more chromosomes have a double strand break. The fragments that are broken off may rejoin to their original chromosome or to a different broken chromosome. In the case of DS, the q22 end of chromosome 21 was translocated to a different chromosome. In order for this to cause DS, the chromosome that received q22 from chromosome 21 must end up in the same gametic cell as an intact chromosome 21. In this situation the gamete would have two copies of chromosome 21q22. It has been shown that and extra copy of only 21q22 is necessary to cause Down's syndrome.
There are 127 known genes and 98 predicted genes of chromosome 21 [8]. This paper will discuss copper zinc superoxide dismutase, collagen XVIII/endostatin, Tiam1, and GATA1. CuZnSOD's activity is increased 50% in DS patients due to its overexpression from trisomy 21.
Endostatin levels in the serum of DS subjects is also increased. Tiam1 is also expression and activity is upregulated due to trisomy 21. The GATA1 gene is not found on chromosome 21; however, it is mutated as a result of the trisomy.
Copper Zinc Superoxide Dismutatse
Superoxide dismutase (SOD) is a primary antioxidant enzyme that catalyzes the dismutation of O2•- to H2O2 (reaction 1) [9]. Two forms of SOD are found with in the cell. Copper zinc SOD (CuZnSOD) is found in the cytosol and nucleus, and manganese SOD (MnSOD) is found in the mitochondria. An association between SOD and cancer has been recognized for many years. In 1979, Oberley and Buettner hypothesized that SOD activity was down regulated in cancer cells [9]. All cancer cells have an altered activity of antioxidant enzymes, particularly SOD. While the majority of cancer cells have a decrease in SOD, some do exhibit an increased activity as compared to their normal cell counterpart.
O2•- + O2•- + 2H+ → H2O2 + O2 (1)
MnSOD has been shown to be a tumor suppressor gene. Other studies suggest that CuZnSOD also exhibits tumor suppressive effects. In one experiment, Zhang and colleagues [10] looked at the effect of overexpressing CuZnSOD in human glioma cells. CuZnSOD overexpressing gliomoa cells that had not induced extra GPx (2.4 and 3.5 ratio CuZnSOD:GPx) exhibited a decreased growth rate. All CuZnSOD overexpressing glioma cells had a decreased tumor volume in vivo as well. Through this they showed that CuZnSOD could work to suppress tumor growth [10]. CuZnSOD was also found to inhibit the invasiveness of human tongue carcinomas. Using antisense to CuZnSOD, Muramatsu and colleagues found an increase in invasion and mobility in low invasive cells [11]. These studies argue that overxpression of CuZnSOD causes a tumor suppressive effect in that it decreases tumor growth rate and the ability to metastasize.
Tumor volume and invasiveness are not qualities that are of importance in leukemia. We also know that cancers from different parental cells respond differently to experimental manipulations. Therefore it is important to describe the effect of CuZnSOD in leukocytes. Kato et al. [12] looked at the level of SOD activity in non-DS human leukemia cells. They found that CuZnSOD activity in AML cells was significantly increased as compared to corresponding normal granulocytes; however, there was a decrease in MnSOD activity in the AML cells. Acute lymphoblastic leukemia (ALL) cells did not significantly differ in either CuZnSOD or MnSOD activity levels when compared to its normal lymphocyte counterpart [12]. This study shows that AML, a common cancer found in DS patients, has a 1.7 fold increase in CuZnSOD. Therefore, CuZnSOD was not found to suppress the cancerous phenotype in these cells. This may explain why the incidence of AML is increased so drastically in DS.
Although it is thought that SOD has a protective effect to have a protective effect by scavenging O2•-, just the opposite may be true. It has been shown that normal human cells typically have a 1:1 ratio of SOD to catalase and glutathione peroxidase (GPx). Since Down's syndrome cells only have an increase in CuZnSOD, its ratio to catalase and GPx is 1.5 [13]. An imbalance of the antioxidant enzymes within the cell may lead to an accumulation of H2O2 which causes damage to cellular components (Figure 1). This imbalance has been hypothesized to be responsible for the etiology of Down's syndrome. Several studies have shown that this increase in CuZnSOD may be involved in the neurological abnormalities and cognitive impairment in DS [14,15]. Groner et al. has shown that damage done to neurotransmitters in transgenic CuZnSOD overexpressing mice is similar to that of DS patients [2].
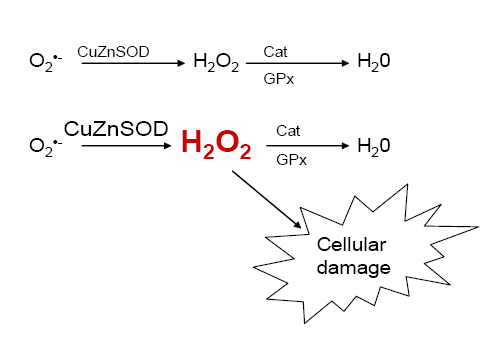 |
Figure 1. Model of antioxidant enzyme imbalance in Down's syndrome cells.
An increase in CuZnSOD but not catalase or glutathione peroxidase will lead to an accumulation of hydrogen peroxide within the cell. This excess hydrogen peroxide may lead to cellular damage. |
CuZnSOD is susceptible to oxidative damage. The excess hydrogen peroxide may attack the very enzyme that formed it. When damaged, Cu(II) is released from the enzyme [16]. The free Cu(II) can be reduced by hydrogen peroxide to form Cu(I) (reaction 2). Via Fenton chemistry Cu(I) and hydrogen peroxide react to form the hydroxyl radical and ion. The Cu(I) also gets oxidized back into Cu(II) and is ready to react with another H2O2 (reaction 3) [17]. The hydroxyl radical is very harmful to the cell and will attack whatever is in its immediate vicinity.
2Cu(II) + H2O2 → 2 Cu(I) + O2 + 2H+ (2)
Cu(I) + H2O2 → 2 Cu(I)OOH + H+ → •OH + OH- + Cu(II) (3)
Midorikawa and Kawanishi [17] showed that CuZnSOD could induce DNA damage in the presense of H2O2. The addition of MnSOD to Cu(II) and H2O2 only increased the DNA damage slightly. However, a large increase in damage occurred in the presence of CuZnSOD. The damage in both cases was dependent on the H2O2 concentration. (Figure 2A). They then looked at DNA damage with increasing amount of CuZnSOD with Cu(II) and H2O2. The addition of CuZnSOD severely damaged the DNA at the lowest concentration (Figure 2B) [17]. These data show that oxidative damage to DNA can be mediated through CuZnSOD. In the excess of CuZnSOD and H2O2, the cell is in danger of being severely harmed.
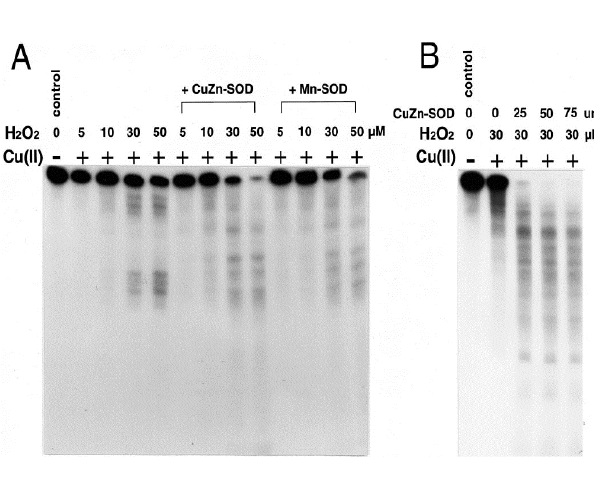 |
Figure 2. DNA damage induced by SOD, hydrogen peroxide, and Cu(II) in vitro.
Autoradiograph of 5' 32P labeled DNA fragment.
A. The DNA was incubated with or without 20 µM CuCl2, 150 U/ml CuZnSOD or MnSOD, and the indicated about of hydrogen peroxide for 20 minutes.
B. The DNA was incubated with or without 20 µM CuCl2 and the indicated amount of hydrogen peroxide and CuZnSOD for 60 minutes [17]. |
It has also been found that DS cells have a diminished ability to repair mitochondrial DNA [18]. At 6 h after treatment with menadione, repair of mitochondrial DNA (mtDNA) damage in DS cells was at 50 to 60% of that of normal cells. Druzhyna et al. speculate that the decrease in repaired mtDNA may be in part due to the increase in CuZnSOD seen in these cells. Oxidative damage through the Fenton reaction, due to an increase in H2O2, may actually increase the initial damage done to the mtDNA in DS cells [18]. If the mitochondrial DNA is not repaired, it could lead to an increase is ROS levels within the mitochondria due to faulty electron transport proteins that are encoded for in the mitochondrial genome. This would lead to a propagation of damage that may assist in the ROS mediated transformation of the cell.
The extra hydrogen peroxide that is being produced within the cells because of increased CuZnSOD activity is not being detoxified. This may cause a prooxidant imbalance within the cell that disrupts or alters cellular processes. Increased levels of hydrogen peroxide have been shown to induce proliferation in normal cells [19], and hydrogen peroxide can act as a second messenger for NF-kB, & AP-1 and cyclic AMP [20]. This taken with the evidence of damage due to •OH, an overexpression of CuZnSOD may in fact lead to a cancerous phenotype.
Collagen XVII and endostatin
The gene for type XVIII collagen is also increased in trisomy 21. Type XVIII collagen works to maintain the integrity of the basement membrane, therefore, the overexpression of type XVIII collagen would help decrease the invasiveness of a tumor [21].
Endostatin, an angiogenesis inhibitor, is found at the C-terminal globular domain of type XVIII collagen [22,23]. This noncollagenous domain is cleaved off by proteases such as cathepsin L or matrix metalloprotease [21]. Serum levels of endostatin in DS patients are increased (38.6 ± 20.1 ng/ml) as compared to non-DS subjects (20.3 ± 11.5 ng/ml) [24]. Endostatin works as an antiangiogenic factor by inhibiting the activity of vascular endothelium growth factor (VEGF) [22,23]. It does so by binding to the tyrosine kinase receptors on VEGF and thus blocking its stimulation [25]. Also, endostatin has been shown to induce apoptosis in endothelial cells in vitro [23].
When a tumor reaches a certain size, the cells in the core become hypoxic. This stimulates the accumulation of the transcription factor hypoxia inducible factor-1 (HIF-1). HIF-1 can induce the expression of many hypoxia related genes, one of which is VEGF [26]. VEGF and other angiogenic factors are upregulated to stimulate the growth of new blood vessels to vascularize the tumor. If the tumor is unable to become vascularized, it will not be able to grow in size. In 1971, Folkman determined that angiogenesis to a tumor was necessary for its growth and for metastasis [27]. Together type XVIII collagen and endostatin work to inhibit the process of invasion and angiogenesis.
Sund et al. [22] showed that a 1.6 fold increase in endostatin levels (similar to that found in DS patients) in transgenic mice was able to reduce the rate of tumor growth in vivo by 3-fold (Figure 3A). Then, they correlated this suppression of tumor growth with the decrease in blood vessels in the tumor (Figure 3B) [22]. Stimulation for angiogenesis is often thought of being a balance between inhibitors and activators. Sund showed that by tipping the balances in favor of antiangiogenic factor, tumor growth could be modulated [22].
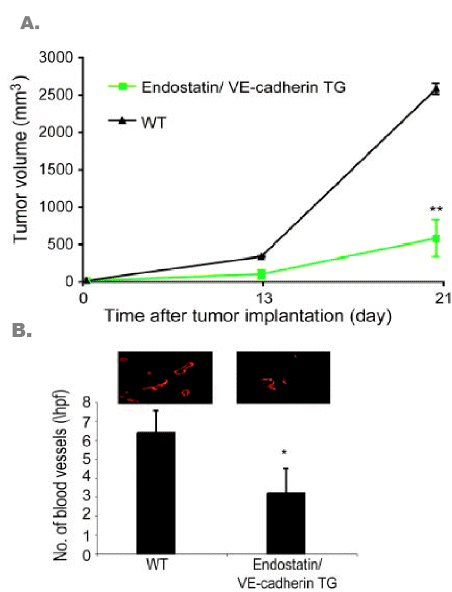 |
Figure 3. Tumor suppression in endostatin transgenic mice.
A. Implated tumor volume growth in endostatin/VE-cadherin overexpressing and wild-type mice.
B. Number of blood vessels found in implanted tumors of the double transgenic and wild-type mice. Endothelial cells of the blood vessels were stained with CD31, an endothelial cell marker [22]. |
Tiam1
Another gene found of chromosome 21q22 is Tiam1. Tiam1 (T-lymphoma invasion and metastasis) is a specific guanine nucleotide exchange for Rac. Rac is a Rho-like GTPase that is activated when it is bound to GTP and inactive when bound to GDP [28]. If Tiam1 is overexpressed due to trisomy 21, then Rac will be active more often, leading to an upregulation of Rac activity. Rac is a downstream signaling molecule of Ras that is involved in cell motility by cytoskeleton rearrangement, cell cycle progression, and in transcription. Rac has also been noted to be a part of cellular transformation caused by Ras. In this way, Tiam1 can be considered an oncogene that is upregulated in subjects with Down's syndrome [28]
GATA1
GATA1 is affected by trisomy 21, however it is not found on the 21st chromosome. GATA1 is encoded for on the X chromosome, and trisomy 21-specific mutations in this gene have been identified. Down syndrome patients have a mutation in the transcriptional activation domain of GATA1 [29]. The exact cause of the mutations is not known; however, two transcriptional cofactors of GATA1, RUNX1 and ETS2, are encoded for on chromosome 21q22 and are postulated to play a role in this mutation [3,29]. GATA1 is a hematopoietic transcription factor that helps with the differentiation of megokaryocytes [3,29]. Therefore, megakaroblasts with this mutation do not develop correctly, although they do proliferate.
Nearly all Down's syndrome children with AMKL have a mutation in GATA1. Interestingly, no non-DS leukemia patients have been found to have this mutation. However, non-Down's syndrome children who acquire a trisomy 21 karyotype due to leukemia may get a mutation in GATA1 [29]. There also is evidence that these mutations occur in utero, accounting for cases of transient leukemia in newborns with Down's syndrome [29]. Mutated GATA1 has been postulated to be an initiating event fr leukemia [29]. Therefore, other factors are needed to get the cancerous phenotype. The imbalance of CuZnSOD could be the promoting event that leads to carcinogenesis of megakaryoblasts in DS subjects.
Experimental hypotheses
CuZnSOD has been shown to have a tumor suppressive effect. However, these studies have been done in already transformed cells [10,11]. It has also been shown an abundance of CuZnSOD can cause damage to the cell [17]. Studies need to be done to tease out whether or not CuZnSOD has a causative role in leukemia progression due to increased ROS production or a protective role against solid tumors in Down's syndrome patients.
In vitro transformation of Down's syndrome cells
This paper hypothesizes that the overexpression of CuZnSOD is acting to damage cellular component, as shown by Midorikawa [17], therefore increasing the rate of carcinogenesis. To test this we propose to try to transform an in vitro culture of Down's syndrome fibroblasts. To determine whether CuZnSOD or endostatin help facilitate or deter the transformation process, RNA interference (RNAi) will be used to knock-down their expression. Antisense adenoviruses to these genes could also be used, however, RNAi gives the advantage because its effect can be permanent and reinfection every few days would not be necessary. Clones of the knockdown cells would be analyzed for relative gene expression using real-time PCR. Clones that expressed CuZnSOD or endostatin at levels comparable to of non-DS fibroblast would be selected. Real-time PCR could also be used on a few genes encoded on chromosome 21q22 to verify that these cultured cells still overexpressed the other DS genes.
To test for transformation frequencies, wild-type cells, CuZnSOD KD, endostatin KD, and CuZnSOD/endostatin KD cells would be immortalized with E6/E7. Normal human cells are not easily transformed and the immortalized cells could help ease the process, as well as, extend the life of the cultures. The cells that were successfully immortalized would be selected for and given a promoting event such as ionizing radiation. The cells would be irradiated with 2 Gy of γ-irradiation and allowed to recover for a week. The surviving cells would then receive a second dose. Four doses of irradiation would be given in this manner. Other types of cancer promoting treatments, such as TPA, could be used instead of irradiation.
At the end of the irradiation scheme, the cells would be checked for transformation using the soft agar assay. Transformed cells exhibit anchorage independent growth and are able to form colonies in soft-agar, whereas, nontransformed cell should not be able to. By comparing the transformation frequencies of the cells, one should be able to tell if CuZnSOD or endostatin are important in carcinogenesis of DS cells. I would expect to see about an equal transformation frequency between the wild-type DS and endostatin KD cells. Endostatins involvement in cancer is probably not promoting and would not have an effect in vitro. A decrease in the transformation frequency of the CuZnSOD KD and CuZnSOD/endostatin KD cells would be expected. This would show that the excess CuZnSOD would help facilitate the transformation of the cell. If the transformation frequency of the cells with decreased CuZnSOD was increased, then it could be argued that CuZnSOD had a tumor suppressive effect. If this experiment was successful, other cell types may be tried to confirm that this phenomenon was not limited to the DS fibroblasts.
It would then, be necessary to determine if to demonstrate the antioxidant imbalance within the DS cell may be causing the transformation of DS cells. To test this, GPx or catalase would be knocked down in normal human fibroblast. Real-time PCR would again be used to determine the levels GPx, catalase, and CuZnSOD expression. Clones that exhibited a 1.5 CuZnSOD:GPx or CuZnSOD:catalase ratio would be selected. These clones would mimic the antioxidant imbalance within the DS cells.
The clones would then be immortalized and treated with radiation as previously described. I would expect that the GPx KD and catalase KD cells would have a slightly higher transformation frequency as compared to wild-type cells. If this were the case, then it would show that the excess H2O2 due to an antioxidant imbalance would be carcinogenic. Hydrogen peroxide levels would be determined to verify that they were increased in the cells.
These experiments would determine whether or not CuZnSOD acts a tumor suppressor or a tumor promoter under these circumstances.
In vivo test of solid tumor formation
It has previously been shown that tumor growth is inhibited in transgenic mice overexpressing endostatin at a level comparable to DS [22]. We believe that the decrease in solid tumor occurrence is due to excess endostatin and inhibition of angiogenesis, and not the inhibition of cellular transformation. Therefore, solid tumor incidence may not be decreased, but rather undetectable. To test this, we propose making double transgenic mice that overexpress CuZnSOD and endostatin, as well as, transgenic mice that overexpress one or the other. The mice will be allowed to live normal lives until they are late in age. At this point they will be screened for cancer. Positron emission tomography (PET) scans will be preformed on the mice to detect any sign of a tumor. F-DG, a radiolabled analog of glucose, is taken up more readily by cancerous cells. Small tumors that are normally undetectable will "light up" on the PET scan. The animals will be sacrifice and histological sections of their tissues will be taken to determine whether a cancerous growth is found where the PET scan indicated one.
The mice overexpressing CuZnSOD are expected to have a higher incidence of tumor than wild-type mice. Because these transgenic mice are not overexpressing endostatin their tumors may be detectable to the naked eye. Wild-type mice may also be bearing tumors. The double transgenic mice would be expected to have the same number of tumors as the CuZnSOD only overexpressing mice. However, because of the excess endostatin, the tumors on the double transfectant mice would be extremely small and otherwise undetectable. These techniques could help determine whether it is the transformation event or just the tumor growth that is inhibited in Down's syndrome.
Conclusions
Down's syndrome is a very prevalent disorder with many manifestations arising from trisomy 21. The overexpression of the genes found on chromosome 21 has a profound effect on the cellular processes within these individuals. Epidemiological studies have shown a large increase in the incidence of leukemia and a decrease in solid tumors in these individuals. It seems almost paradoxical that DS patients have such a high increase in leukemia and a decreased occurrence of solid tumors. This paper proposes that the transformation events throughout the body are actually increased in DS subjects; however, due to excess endostatin these cancers can not grow into a tumor that is of detectable size. Since leukemias are circulating cancers, the inhibition of angiogenesis does not affect them.
Leukocytes in DS patients have a mutated GATA1 gene that is thought to be an initiator for carcinogenesis [29]. The increase in ROS due to an antioxidant imbalance may then act as a promoting event, thus making the overexpression of CuZnSOD a damaging agent to the cell. Leukocytes in DS patients also overexpress Tiam-1 which activates Rac in the Ras pathway [28]. This could also help explain why leukemia is increased. However, more research is needed to determine the role of CuZnSOD in cancer of Down's syndrome patients.
References
- Massey GV. (2005) Transient leukemia in newborns with Down syndrome. Pediatr Blood Cancer. 44: 29-32.
- Groner Y, Elroy-Stein O, Avraham KB, Schickler M, Knobler H, Minc-Golomb D, Bar-Peled O, Yarom R, Rotshenker S. (1994) Cell damage by excess CuZnSOD and Down's syndrome. Biomed Pharmacother. 48: 231-240.
- Hitzler JK, Zipursky A. (2005) Origins of leukaemia in children with Down syndrome. Nat Rev Cancer. 5: 11-20.
- Hasle H. Clemmensen IH, Mikkelsen M. (2000) Risks of leukaemia and solid tumours in individuals with Down's syndrome. Lancet. 355: 165-169.
- Satgé D, Sommelet D, Geneix A, Nishi M, Malet P, Vekemans M. (1998) A tumor profile in Down syndrome. Am J Med Genet. 78: 207-216.
- Mann JR, Pearson D, Barrett A, Raafat F, Barnes JM, Wallendszus KR. (1989) Results of the United Kingdom Children's Cancer Study Group's malignant germ cell tumor studies. Cancer. 63: 1657-1667.
- Satgé D, Sasco AJ, Carlsen NL et al. (1998) A lack of neuroblastoma in Down syndrome: a study from 11 European countries. Cancer Res. 58: 448-452.
- Hattori M, Fujiyama A, Taylor TD et al. (2000) The DNA sequence of human chromosome 21. Nature. 405: 311-319.
- Oberley LW, Buettner GR. (1979) Role of superoxide dismutase in cancer: a review. Cancer Res. 39: 1141-1149.
- Zhang Y. Zhao W. Zhang HJ, Domann FE, Oberley LW. (2002) Overexpression of copper zinc superoxide dismutase suppresses human glioma cell growth. Cancer Res. 62: 1205-1212.
- Muramatsu H. Kogawa K, Tanaka M, Okumura K, Nishihori Y, Koike K, Kuga T, Nitsu Y. (1995) Superoxide dismutase in SAS human tongue carcinoma cell line is a factor defining invasiveness and motility. Cancer Res. 55: 6210-6214.
- Kato M, Minakami H, Kuroiwa M, Kobayashi Y, Oshima S, Kozawa K, Morikawa A, Kimura H. (2003) Superoxide radical generation and Mn- and Cu-Zn superoxide dismutases activities in human leukemic cells. Hematol Oncol. 21: 11-16.
- Sinha S. (2004) Anti-oxidant gene expression imbalance, aging and Down's syndrome. Life Sci. 76: 1407-1426.
- Ceballos-Picot I, Nicole A, Briand P, Grimber G, Delacourte A, Defossez A, Javoy-Agid F, Lafon M, Blouin JL, Sinet PM. (1991) Neuronal-specific expression of human copper-zinc superoxide dismutase gene in transgenic mice: animal model of gene dosage effects in Down's syndrome. Brain Res. 552: 198-214.
- Thiel R, Fowkes SW. (2005) Can cognitive deterioration associated with Down syndrome be reduced? Med Hypoth. 64: 524-532.
- Li Y, Kuppusamy P, Zweir JL, Trush MA. (1996) Role of Cu/Zn-superoxide dismutase in xenobiotic activation. II. Biological effects resulting from the Cu/Zn-superoxide dismutase- accelerated oxidation of the benzene metabolite 1,4-hydroquinone. Am Soc Pharmacol Exp Therapeut. 49: 412-421.
- Midorikawa K, Kawanishi S. (2001) Superoxide dismutases enhance H2O2-induced DNA damage and alter its site specificity. FEBS Lett. 495: 187-190.
- Druzhyna N, Nair RG, LeDoux S, Wilson GL. (1998) Defective repair of oxidative damage in mitochondrial DNA in Down's syndrome. Mut Res. 409: 81-89.
- Laurent A, Nicco C, Chéreau C, Goulvestre C, Alexandre J, Alves A, Lévy E, Goldwasser F, Panis Y, Soubrane O, Weill B, Batteux F. (2005) Controlling tumor growth by modulating endogenous production of reactive oxygen species. Cancer Res. 65: 948-956.
- Gupta A. Rosenberger SF, Bowden GT. (1999). Increased ROS levels contribute to elevated transcription factor and MAP kinase acitivities in malignantly progressed mouse keratinocyte cell lines. Carcinogenesis. 20: 2063-2073.
- Fakai N, Eklund L, Marneros AG, Oh SU, Keene DR, Tamarkin L, Niemalä M, Li E, Pihlajaniemi T, Olsen BR. (2002). Lack of collagen XVIII/endostatin results in eye abnormalities. EMBO J. 21: 1535-1544.
- Sund M, Hamano Y, Sugimoto H, Sudhakar A, Soubasakos M, Yerramalla U, Benjamin LE, Lawler J, Kieran M, Shah A, Kalluri R. (2005) Function of endogenous inhibitors of angiogenesis as endothelium-specific tumor suppressors. PNAS. 102: 2934-2939.
- Sasaki T, Larsson H, Tisi D, Claesson-Welsh L, Hohenester E, Timpl R. Endostatins derived from bollagens XV and XVIII differ in structural and binding properties, tissue distribution and anti-angiogenic activity. J Mol Biol. 301: 1179-1190.
- Zorik TS, Mustacchi Z, Bando SY, Zatz M, Moreira-Filho, Olsen B, Rassos-Bueno. (2001) High serum endostatin levels in Down syndrome: implications for improved treatment and prevention of solid tumours. Eur J Human Gen. 9: 811-814.
- Yao YG, Duh EJ. (2004) VEGF selectively induces Down's syndrome critical region 1 gene expression in endothelial cells: a mechanism for feedback regulation of angiogenesis? Biochem Biophys Res Comm. 321: 648-656.
- Richard DE, Berra E, Pouysségur J. (1999) Angiogenesis: how a tumor adapts to hypoxia. Biochem Biophys Res Comm. 266: 718-722.
- Folkman J. (1971) Tumor angiogenesis: therapeutic implications. New Engl J Med. 285: 1182-1186.
- Mertens AE, Roovers RC, Collard JG. (2003) Regulation of Tiam 1-Rac signaling. FEBS Lett. 546: 11-16.
- Crispino JD. (2005) GATA1 mutations in Down syndrome: implications for biology and diagnosis of children with transient myeloproliferative disorder and acute megakaryoblastic leukemia. Pediatr Blood Cancer. 44: 40-44.