Professor Dr. G. Lubec, CChem, FRSC (UK)
Ephrem Engidawork
J Neurol 2002 249(10):1347-56
Department of Pediatrics, University of Vienna
Währinger Gürtel 18-20
1090 Vienna, Austria
Tel.: +43-1/404003215
Fax: +43-1/404003194
|
 |
Reprinted with permission of Thomas Tschech, M.A.
Herstellung/Production
Dr. Dietrich Steinkopff Verlag GmbH & Co. KG
Poststraße 9, 64293 Darmstadt
Germany
Tel.: +49-6151-82899-19
Fax: +49-6151-82899-40
|
Abstract
Down syndrome (DS) is the most common genetic birth defect associated with mental retardation. The mechanism(s) underlying the neuropathology of DS is not completely understood. Different hypotheses have been advanced to explain this mystery, including the gene dosage effect, the amplified developmental instability, and the molecular misreading concept. Overexpression of genes residing in chromosome 21 has been assumed to be a central point in the neuropathology of DS, although reports disagreeing with this notion have also been published. In addition, an accumulating body of evidence indicates that genes located on other chromosomes are also involved in the process. DS thus appears to be a disease process involving numerous gene products and this interaction and interplay in the final analysis determines the outcome of the disease. In this regard, transcription factors, reactive oxygen species and apoptosis related proteins are viewed as potential candidates that play a significant role in the disease process. Therapeutic modalities that target these factors including antioxidants and caspase inhibitors might have some benefit in alleviating the symptoms of DS.
Key words
Down syndrome · transcription factors · oxidative stress · apoptosis · superoxide dismutase · brain · caspases
Introduction
Down syndrome (DS) is the most common human genetic disorder that occurs at a rate of 1 in 700-800 live births. More than 99% of the individuals with DS have an extra copy of the entire chromosome 21, 95% of which are free trisomies and the remainder are mosaics or have translocations. DS involves dysmorphic features, which collectively constitute its distinctive physical phenotype, endocardial and immunological defects, hematological and endocrinal alterations, behavioral and cognitive deficits, mental retardation, and hypotonia, [21]. In addition, numerous studies have documented the presence of senile plaques and neurofibrillary tangles, hallmarks of Alzheimer's disease (AD).
Although most of the aforementioned clinical features of DS are often inconsistent in their occurrence, mental retardation remains the invariable hallmark of this disorder [21]. The nature of the brain defect, however, has not yet been fully elucidated and no consistent specific pathological finding has been described so far. Several hypotheses that range from the gene dosage effect through the amplified developmental instability to the molecular misreading concept [21,60,70,77] have been put forward to explain the neuropathological changes observed in this disorder. However important these hypotheses for basic principles and research concepts of DS may be, we designed this review to give further insight into the molecular processes underlying AD neuropathology based on published data and discuss their relevance to one of the hypotheses when applicable.
The neuropathological changes
DS brains are characteristically small, rounded, foreshortened, and exhibit a steep rise of the occipital lobes, extreme narrowing of the superior temporal gyri, incomplete opercularization with exposure of the insular cortex, and reduced secondary sulcal development [17]. These abnormalities are largely due to diminished and malformed growth of the frontal and temporal lobes secondary to impaired neuronal differentiation [43]. Others share this view [11,15] and add that brain weight
is usually in the low normal range and brain stem and cerebellum are small in relation to the cerebral hemispheres. Moreover, the anterior commissure in adults with DS is reduced in cross-sectional area [75]. All these macroscopical findings, however, are neither absolutely specific nor consistent [67].
Histological changes observed in DS brain include
abnormalities in cortical lamination, irregular clustering of neuronal cell bodies, muted dendritic arborization and proliferation of dystrophic neurites. This cortical neuritic pathology is associated with increased phospho-tau and consistently accompanied by aberrant
expression of genes that are normally modulated with
neuritic growth during development, regeneration and
degeneration, including growth associated protein (GAP-43), neuronal thread protein (NTP) and nitric oxide synthase 3 (NOS3) (Fig. 1) [17]. Microglia are prominent and vascular dysplasias with focal calcification in
basal ganglia can be found [17]. Nerve cell heterotopias in the white layers of the cerebellum indicate disturbance of embryonic cell migration [61]. Abnormalities in morphology and number of dendritic spines including atrophy of the dendritic tree of visual cortex have been described [5,6,52,76]. Other reported defects in brain histogenesis are poverty of granular cells throughout the cortex, decreased neuronal densities in layers II and IV of the occipital cortex [81], and diminution in the number of hypothalamic neurons [80]. There is also evidence for abnormalities of neuronal differentiation and abnormal migration in fetal and infant brain [21]. Neuronal loss is prominent from birth onwards [11,14,82]
but a consistent picture of the "wiring" of the brain in DS has not yet emerged.
| Fig. 1 Neuritic and glial sprouting in brains with DS: detected with monoclonal antibodies to phospho-neurofilament (A), neuronal thread protein (B1,B2), nitric oxide synthase3, NOS3 (C,D),and the growth associated protein, GAP-43 (E,F). Immunoreactivity was revealed with the avidin-biotin horseradish peroxidase labeling system using diaminobenzidine as the chromogen (dark brown precipitate). The sections were counterstained with hematoxylin to provide a light blue contrast for unlabelled structures. Neuritic sprouts are vesicular (A) or coarse-granular to punctate in morphology (B,C,E). Glial sprouts are mainly punctate and often juxtaposed to glial cell nuclei (D,E). Courtesy of Dr. de la Monte. |
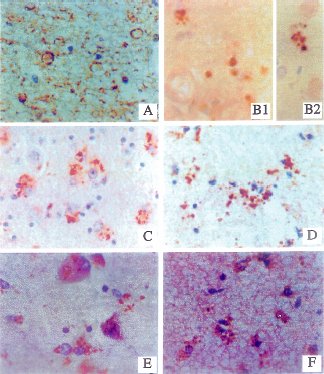 |
It is of clinical interest to note that virtually all DS patients develop AD-like pathological changes by thefourth decade of life [50,66]. Accumulation of senile
plaques, beta amyloid deposits and neurofibrillary tangles occur from that time point, representing or may be leading to AD neurodegeneration. Neurohistological
features, however, are complex and variable. Although it
is accepted that DS patients show AD-like neuropathological changes, there are subtle differences between DS and AD histopathology [11,51].
What leads to the neuropathological changes?
It is becoming increasingly clear that the overall DS abnormality is caused by altered expression of more than
a single gene. Indeed, the process involves a multitude of
gene products and the interplay between these products
appears to determine the outcome of DS.
Transcription factors/protooncogenes
We realize from neuropathological findings that the abnormal molding of brain in DS is a molecular misconception resulting from a serious deterioration of the
concerted action of factors normally modeling and
wiring the brain. The major factors for the complex
structuring of the brain are thousands of transcription
factors (TFs) which wax and wane during different
phases of brain development and wiring, thus enabling
the required mass of information necessary for histoarchitectonics. TFs are not only key regulators of building the brain, but also are central in cross-talks among cells
and signaling molecules. TFs for e.g. homeobox genes, TFs with the POU domain, helix-loop-helix motifs, leucine-zippers and forkhead genes, determine proliferation, programmed cell death and differentiation of neurons and glial cells and thus mainly control and contribute to forming the brain. The reader is referred to the excellent reviews on this intriguing subject [3,33].
Although a series of TFs are encoded on chromosome 21 [9,12,13], no systematic quantification of these TFs has been carried out in DS brain. Thus, to gain an
understanding of how extra gene load affects TFs encoded on chromosome 21, the expression of Ets-2 was investigated. Ets-2 is a proto-oncogene and TF ubiquitously expressed and involved in organogenesis. It is encoded on genes residing in the so-called "critical region"
of chromosome 21. The expression of Ets-2 was determined by RT-PCR in several brain regions of controls and DS patients as well as in heart biopsies obtained following surgery [27,28,44].
Surprisingly, Ets-2 was not over-expressed in any of
the brain regions examined, but significantly decreased
in frontal and temporal lobes of patients with DS. In
heart, Ets-2 levels were comparable between DS and
controls. This finding challenged the over-expression
hypothesis by indicating that over-expression per se can
not fully account for the DS phenotype. Indeed, given
the role of Ets-2 in organogenesis, over-expressed Ets-2
would have been able to explain the dysmorphic features, brain, heart and immunological pathologies, and
last but not least the increased leukemia rate found in
DS. The deranged Ets-2 in our studies point to an involvement of this TF in the pathomechanisms of DS and
Ets-2 deterioration would also interfere with plasticity
of the brain in all age groups.
Ets-family members appear to require co-operation
with other factors for their activity (Fig. 2). Ets-2 absolutely requires Fos and Jun for activation of the oncogene-responsive unit of the polyoma virus enhancer [79] and the Ets-like motif of the Fos promoter is required in conjunction with the Fos-AP-1 motif for induction of promoter activity [30].
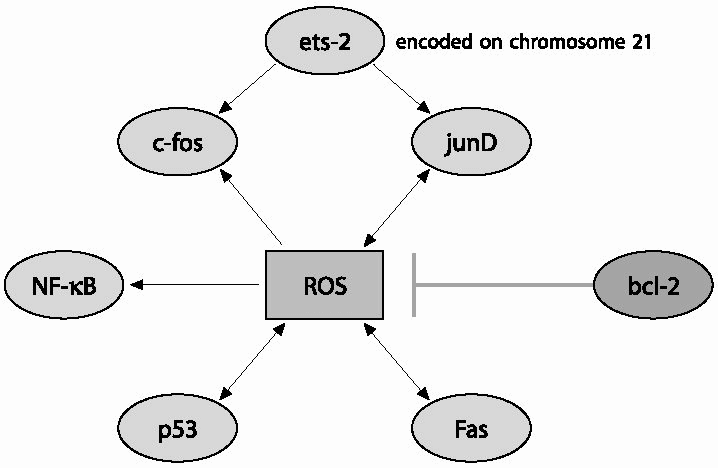 |
| Fig. 2 Interaction of reactive oxygen species (ROS) with transcription factors (TFs): Ets-2 a TF encoded on chromosome 21 may not interact directly with ROS, but as it co-operates with the AP-1 TFs (c-Fos and JunD) this co-operation probably provides a link between Ets-2 and ROS. All the other TFs are capable of interacting directly with ROS by which one modulates the function of another. Oxidative stress leads to an increase in c-Fos expression,an event linked to apoptosis and reduced synaptic plasticity,whereas decreases JunD, a protein that up-regulates antioxidant response element (ARE) mediated expression and coordinated induction of detoxifying enzymes in response to oxidative insults. ROS can increase the expression of p53 and Fas. Likewise, p53 and Fas can lead to increased ROS production indicating a feed-forward mechanism to ensure death of the cell committed to die. ROS activate cytoplasmic NF-K B by inducing phosphorylation and degradation of the inhibitory NF-K B proteins. Following this activation, NF-K B is translocated into the nucleus and binds K B sequences on its target genes to modulate their expression. Unlike the other TFs, Bcl-2 displays antioxidant properties by acting as a non-reactive free radical scavenger. |
Gene hunting for aberrant gene expression using
subtractive hybridization in fetal DS brain has indeed
revealed down-regulation of a sequence with 100% homology with the TF JunD. Studies at the protein level
showed that JunD was decreased in temporal, frontal
cortex and cerebellum of patients with DS [46]. The Jun
family of TFs or transcription regulator proteins, are
components of the AP-1 transcription factors, which
consist of three members: c-Jun, JunB and JunD. These
proteins share a high degree of homology at the primary
sequence level but are functionally different. It has recently been shown that mouse JunD negatively regulates fibroblast growth and antagonizes transformation by
the proto-oncogene Ras [59]. The study clearly demonstrated that c-Jun decreases while JunD accumulates when fibroblasts become quiescent;when resting cells
were stimulated, nuclear localized JunD was rapidly degraded. Over-expression of JunD resulted in slower growth and an increase in the percentage of cells in
G0/G1, whereas c-Jun over-expression produced larger
S/G2 and M phase populations. JunD furthermore partially suppressed transformation of cells by activated TF Ras, whereas c-Jun co-operated with Ras to transform
cells. The finding of reduced JunD in DS brain may be of
relevance for the pathological findings given above as
the TF AP-1 was shown to be involved in neurogenesis
[41,58]. Moreover, the association of JunD with other
nuclear TFs to up-regulate mechanisms critical to cellular protection against oxidative stress [78] argues for the importance of this TF in DS pathology.
The aforementioned interaction between Fos and
Ets-2 along with the importance of Fos in plasticity and
long-term potentiation, the experimental basis for cognitive functions such as memory and learning [38,62], prompted us to investigate c-Fos levels in DS brain by
RT-PCR [26]. We found a significantly and remarkably
increased c-Fos mRNA steady state in frontal, parietal
and temporal cortices of DS brain. This observation can
be linked to impaired plasticity, overexcitation [32], apoptosis, oxidative stress [10], and to the impairment
of the concerted action of TFs in DS brain.
Taken together, the presence of a large number of TFs
with diverse sequence-specific interactions with DNA
sites and amongst themselves, and their ability to be
modified in response to a variety of environmental cues
and intracellular signals, provide combination codes for
highly complex and yet highly organized patterns of
gene expression that are likely to determine the diversity
of neuronal phenotypes. The shown impairment of this
TF system may reasonably explain the molecular biological basis for the maldevelopment and pathology of DS brain.
Reactive oxygen species
Reactive oxygen species (ROS) are compounds that have a lone electron in their outer orbit. This electron configuration confers to the compound an unusual reactivity. As a result, ROS have a tendency to attack neighboring molecules in reactions that can be propagated in an autocatalytic manner. The important production sites of
such ROS (e.g. the superoxide (.O2-) and hydroxyl (.OH) species) are the mitochondrial respiratory chain and the sequences catalyzed by cyclo-oxygenases and lipo-oxygenases.
The link between TFs/proto-oncogenes and reactive oxygen species (ROS) in general has been addressed previously [54]. More specifically, TFs for e.g. Fos (see above) and nuclear factor kappa B (NF-kB), [40] deranged in DS brain [45], have been shown to strongly interact with the redox system (Fig. 2). And this is a checkpoint where molecular biology meets ROS-biochemistry.
Considerations that ROS are involved in the pathogenesis of DS date back two decades [21,71,72]. A milestone in this development was the concept that SOD1, encoded by the so-called DS critical region [70] on chromosome 21 was over-expressed as a result of gene
dosage effect [2,21]. The gene coding for SOD1 is localized to chromosomal region 21q22.1. SOD1 catalyses dismutation of superoxide anions to hydrogen peroxide, which in turn is detoxified by glutathione peroxidase (Gpx) and catalase. This two-step process leads to the elimination of hydrogen peroxide and other noxious radicals. If SOD1 is over-expressed without a concomitant increase in Gpx and catalase, increased hydrogen peroxide capable of inducing apoptosis per se [57] and potentially generating the highly reactive hydroxyl radical [49] would be produced, owing to the disturbance between the two steps of the antioxidant pathway. There is now evidence available that shows Gpx and catalase levels do not increase to compensate elevated SOD1 expression in DS brain and subsequently increased hydrogen peroxide tissue levels could be expected and indeed have been shown [16].
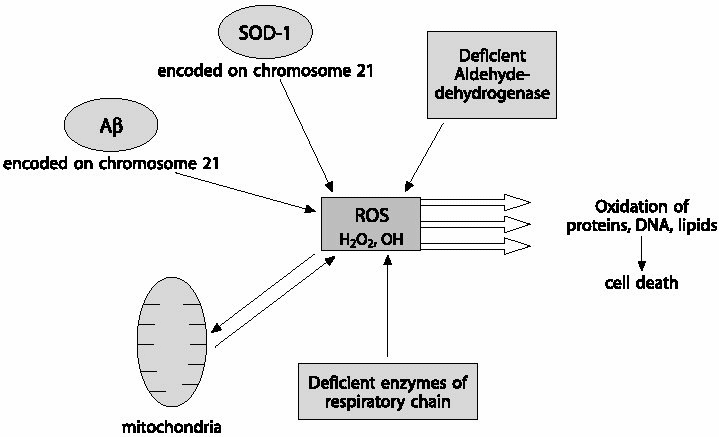 |
| Fig.3 Possible sources of reactive oxygen species (ROS): ROS are normally produced in the living cells, the major source being the mitochondria. The shift in the equilibrium between the oxidant and antioxidant pathway to oxidant production, results in oxidative stress. This usually occurs under pathological conditions such as DS. Increased expression of SOD-1 in DS leads to over-production of hydrogen peroxide that is too large to be handled by the processing enzymes. Thus, high levels of hydrogen peroxide would be produced which leads to cell damage directly or through production of the more noxious hydroxyl radical. A ß, one of the neuropathological products of DS is also capable of producing ROS by itself. Decreased aldehyde dehydrogenase activity, another biochemical feature of DS, could produce ROS, as it is the major enzyme involved in detoxifying endogenously produced reactive carbonyls and aldehydes. ROS produced in such a way coupled to deficiency in enzymes of the electron transport chain may further compromise the mitochondrial respiration, leading to amplified generation of free radicals that ultimately lead to cell death. |
Although there is accumulating evidence that RO play a key role in DS, the mechanism(s) leading to generation of ROS has not yet been fully elucidated. There is no proof that increased SOD1 leads to increased lipid peroxidation in DS as described by several authors [35]. Although several groups have published data on lipid peroxidation, this may not be derived from increased SOD1, rather from mitochondrial damage or defects in electron transfer chain enzymes and aldehyde dehydrogenase in DS (Fig. 3) [42,48]. On the other hand, increased SOD1 (whose activity as protein expression was never systematically studied) could be an innocent bystander. We addressed the possibility that increased SOD1 is a secondary phenomenon simply brought into play to detoxify increased superoxide anion caused by a mechanism different from primary over-expression of SOD1 [29]. Using a proteomic approach that unambiguously identified and quantified SOD1 and SOD2 concomitantly, it was revealed that in brain of patients with DS, only SOD1 encoded on chromosome 21 but not SOD2 encoded elsewhere, was over-expressed. If both SODs were increased, one would have suggested that these superoxide dismutating enzymes were increased secondarily to an increase in superoxide anion. However attractive the proposed over-expression of SOD1 is, it finally remains open whether SOD1 is the underlying cause or simply an epiphenomenon. But evidence favors the former: moderately increased SOD1 in transgenic mice generated significant brain damage [23]. Even if elevated SOD1 levels were a secondary phenomenon, the primary or secondary involvement of ROS in DS pathology is highly probable. It has been shown that fetal DS cortical neurons in vitro generate increased levels of ROS, degenerate and undergo apoptosis. The extent of degeneration and apoptosis of these neurons could be diminished by the application of free radical scavengers [10].
Transcription factors, reactive oxygen species and apoptosis-closing the circle
ROS modulate TFs and TFs modulate ROS generation; the consequence of any imbalance may be neuronal death and neurodegeneration in DS. What is the nature of the interaction between TFs and the redox system? One example for understanding of how oxidation/reduction reactions modify TFs is the Fos and Jun system: Fos and Jun proteins form a heterodimeric complex that interacts with the DNA regulatory element known as the activator protein 1 (AP-1) binding site. Studies indicate that oxidative modification of a single conserved cysteine residue in the DNA binding domain of the two proteins significantly alters transcriptional activity mediated by AP-1 binding factors [1]. Based on this finding and others, it has been suggested that redox regulation could be a general mechanism of control for TFs [1]. Another model of how a TF interacts with the redox system is given by bcl-2 functions in an antioxidant pathway. Bcl-2 (Fig. 2), which is localized to mitochondria, endoplasmic reticula and nuclear membranes and protects cells from hydrogen peroxide and menadione-induced oxidative death and suppresses lipid peroxidation. Bcl-2 also protects cells from gamma-irradiation [73], the most potent source for free radical generation. Over-expressed bcl-2 completely inhibited cell death from as much as 0.5 mM hydrogen peroxide [34]. Studying bcl2 expression in DS brain, recent data clearly showed aberrant bcl-2 expression [64,65], a finding confirmed in our studies [20]. Excess hydrogen peroxide levels may well be responsible for increased bcl-2 expression in AD brain, if bcl-2 in DS is not up-regulated secondarily by other TFs. Either way Bcl-2 upregulation in DS can be viewed as a response elicited by surviving neurons to protect themselves from an eerie of oxidative stress.
| Fig. 4 Increased NOS3 (pale regions within structures)immunoreactivity associated with increased expression of the p53 (darkly stained structures) pro-apoptosis gene product in medium size cerebral meningeal vessels and neuropil neurites.(A,B) The p53 immunoreactivity is localized in nuclei of smooth muscle cells, and the NOS3 immunoreactivity is present in the cytoplasm of endothelial and smooth muscle cells. (C,D) Small cortical NOS3-immunoreactive vessels surrounded by p53-positive cell processes suggesting associated degeneration of perivascular glial cell processes. (E) Lack of NOS3 immunoreactivity in a neuritic senile plaque that has abundant p53-immunoreactive (degenerated)dystrophic neurites. (F) Co-localization of NO3 with p53 in dystrophic cortical neurites (all of the dark irregular structures represent neurites that are double-labeled). Courtesy of Dr. de la Monte. |
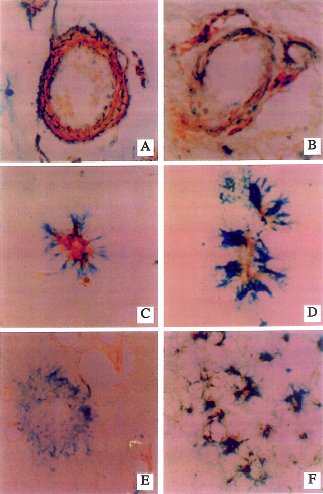 |
Apart from ROS-TFs interactions shown above, ROS
are interacting with a series of TFs and apoptotic factors (Fig. 2), among them is p53, a key protein and transcriptional activator that is considered as the "guardian of the
genome". There is fair evidence showing that p53 is strongly associated with ROS [19,39,47]. p53 is essential for cell cycle arrest and induction of apoptosis in response to chromosomal damage [31,63] but is also involved in cell responses to a variety of insults, such as metabolic deprivation or deranged expression of transcription factors [22]. In DS it is of utmost importance that p53-mediated apoptosis can occur both through transcription dependent and independent mechanisms [63]. Target genes of p53 that are proposed as involved in apoptosis include a cell cycle protein p21/WAF1, the apoptotic protein Bax and the death receptor APO-1/Fas (CD95) [31,56,63]. This provides a link between the cell cycle, known to be deteriorated in DS [8,55], RO and p53, as p53 was found to be over-expressed in DS brain [18,68]. In addition, p53 is co-localized with other gene products that play a role in production of ROS and apoptosis, such as NOS3 and amyloid ß (Aß) (Figs. 4 and 5). [17]. NOS3 is an isoform of NOS that generates nitric oxide (NO) from arginine. The enzyme is constitutively activated, and is expressed in central nervous system neurons [17]. NO by itself is a free radical that can be toxic to neuronal cells. In addition, it can react with superoxide radical to generate a peroxinitrite intermediate that subsequently decomposes to the toxic hydroxyl radical. Aß causes hydrogen peroxide and lipid peroxide accumulation in cells [7] and was also shown to activate caspases by cross-linking the death receptors (Figs. 3 and 6) [36,53]. The aberrant expression of NOS3, Aß and p53 in DS brain argues for their potential role in apoptosis and further provides evidence for the interaction of p53 and ROS. Since apoptosis involves damage to mitochondria, it might lead to generation of ROS, which could further amplify the cycle of events.
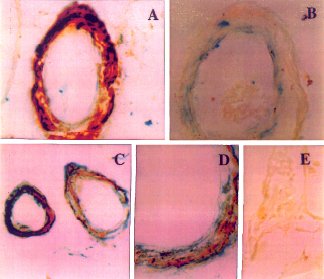
| Fig. 5 Amyloid ß (A ß) angiopathy associated with expression of the p53 pro-apoptosis gene in smooth muscle and endothelial cells of cereberal vessels. (A) Intense levels of A ß immunoreactivity co-localized with p53 in vascular smooth muscle cells of a leptomeningeal vessel.The endothelial cells are positive for p53 but negative for A ß. (B) increased p53 immunoreactivity in a cerebral vessel devoid of A ß immunoreactivity. The dark grey labelling along the inner edge of the blood vessels is positive for p53. (C,D) Co-localization of A ß and p53 immunoreactivity in smooth muscle cells of vessels within the cerebral cortex shown at low (C) and high (D) magnification. The darkly stained regions are positive for p53, and the pale grey regions are positive for A ß. (E) Normal cortical vessel lacking both A ß and p53 immunoreactivity. Courtesy of Dr. de la Monte. |
 |
The association between p53 and CD95 is another strong point in the ROS-pathomechanisms of DS that supports p53 and ROS-involvement. ROS not only are linked to p53 but also to CD95 directly (Figs. 2 and 6) [37,74] and deranged brain CD95 expression in DS has been reported previously [18,68]. CD95 belongs to the tumor necrosis factor/nerve growth factor superfamily of cell-surface proteins and has been shown to mediate receptor-dependent programmed cell death. When engaged by CD95 ligand, CD95 oligomers are formed on
the target cell and a signal to enter the cell's intrinsic apoptotic pathway is transmitted via the cytoplasmic so-called "death domain", which in turn triggers the caspase cascade executing apoptosis (Fig. 6) [4].
Conclusion
As described above and in the Figures (Figs. 3 and 6), we provide evidence for a role of transcription factors and ROS in the pathogenesis of DS based on data obtained from our laboratory and elsewhere. We could add significant material from the literature supporting the involvement of TFs and ROS but for the readability and
because of the length of this review we have to limit evidence and apologize to many important contributors to the subject for not having them cited. We also do not ignore many other facts and findings that may well contribute to the pathogenesis of DS.
In this review it is proposed that ROS derived from over-expression of SOD1, or Aß-mediated release, or mitochondrial damage could lead to abnormal brain development early in life per se and by activating other systems (for e.g. TFs). It is known that ROS interact with
TFs and that TFs per se model the brain in development. Moreover, TFs are classical amplifying molecules in the sense that disturbance in their level can substantially affect the end product whether it be damage or change in gene expression. Thus, we consider TFs as major candidates for abnormal wiring and plasticity of the DS brain. Apoptosis of neurons and glial cells may be responsible for the multitude of pathological findings in the fetus and later in life when Alzheimer's disease like neuropathological findings occurs from the fourth decade
[11]. Enhanced apoptosis in DS brain, can be caused by TFs and ROS (Fig. 6). The biochemical pathways leading to DS neuropathology are not clearly understood. However, it appears that ROS as a trigger and mediator, TFs and Aß as promoters, the pro- and anti-apoptotic proteins as regulators and caspases as effectors all play a role during neuronal death.
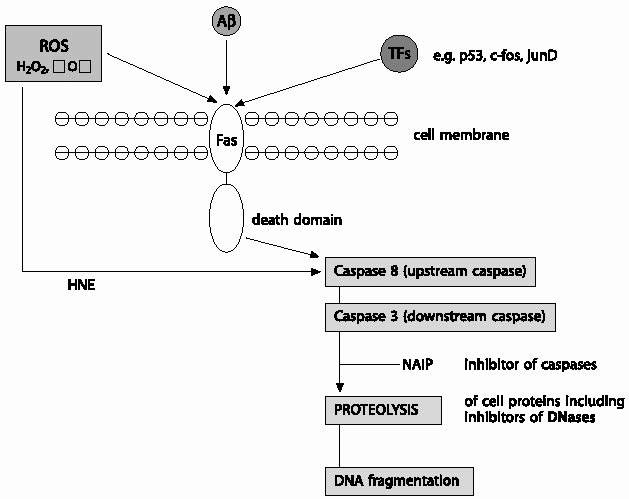 |
| Fig.6 Reactive oxygen species (ROS) and/or transcription factors (TFs) triggered death receptor mediated apoptosis: ROS and/or TFs may increase surface expression of Fas or cause the release of death ligands that leads to the recruitment of a variety of adaptor domain bearing proteins. These adaptor domains glue components of the death machinery together through homophilic interactions and transmit signals from receptors to downstream effectors,such as the caspases. Accordingly, Fas associated death domain (FADD) recruited by Fas, in turn recruits the initiator (upstream) caspases such as caspase-8 to the complex through the death effector domain (DED) module, an event that leads to the proteolytic cleavage of active caspase-8. Active caspase-8 can initiate the caspase cascade by directly cleaving the terminator (downstream) caspases such as caspase-3 that in turn cause proteolytic degradation of a variety of intracellular proteins and DNA fragmentation, eventually leading to cell destruction. ROS, such as 4-hydrox-ynonenal (HNE) can also directly activate caspases independent of CD95 activation leading to oxidative stress induced cell death. Caspase activation can be ablated by inhibitor of caspases, such as neuronal apoptosis inhibitory proteins (NAIP). |
Although we discussed the brain exclusively, we are aware that these pathogenetic principles of TF and RO abnormalities may be extrapolated to other organ systems, such as the heart and dysmorphic features.
Tentative implications for future therapeutic concepts and strategies are obvious: it may well be worth testing antioxidant protocols, as in vitro and in vivo oxidative stress has been repeatedly described in various DS organs. Given apoptosis to be the likely mechanism for neuronal loss in DS, caspase inhibitors are other treatment modalities that deserve attention, as caspases are the ultimate effector of the cell death machinery. Although some innovative concepts for the treatment of the cognitive (serotoninergic and cholinergic) deficit in DS have been tested [24,25,69], there is still therapeutic nihilism in DS. More than a century after the description of DS and with a high incidence in all populations, specific brain research is still far from adequate.
Acknowledgements
The financial assistance for our Down syndrome projects by the Red Bull Company, Salzburg, Austria, is highly appreciated.
References
- Abate C, Patel L, Rauscher FJ, Curran T (1990) Redox regulation of Fos and Jun DNA-binding activity in vitro. Science 249:1157-1161
- Anneren G, Edman B (1993) Down syndrome - a gene dosage disease caused by trisomy of genes within a small segment of the long arm of chromosome 21, exemplified by the study of effects from the superoxide dismutase type 1 (SOD-1) gene. APMIS (Suppl) 40:71-79
- Arenander AT, De Vellis J (1994) Development of the nervous system. In: Siegel GJ, Agranoff BW, Albers RW, Molinoff PB, editors. Basic Neurochemistry, 5th Edition, New York: Raven Press, pp 573-606
- Becher B, Darker PA, Owens T, Antel PJ (1998) CD95-CD95L: can the brain learn from the immune system? Trends Neurosci 21:114-117
- Becker L, Mito T, Takashima S, Onodera K (1991) Growth and development of the brain in Down syndrome. In: Epstein CJ, editor. The Morphogenesis of Down syndrome. New York: Wiley-Liss, p133
- Becker LE, Armstrong DL, Chan F (1986) Dendritic atrophy in children with Down's syndrome. Ann Neurol 20:520-532
- Behl C, Davis JB, Lesley R, Schubert D (1994) Hydrogen peroxide mediates amyloid ß protein toxicity. Cell 77:817-827
- Bernert G, Nemethova M, Cairns N, Lubec G (1996) Decreased cyclin dependent kinase in brain of patents with Down syndrome Neurosci Lett 216:68-70
- Blouin JL, Duriaux-Sail G, Guipponi M, Rossier C, Pappasavas MP, Antonarakis E (1998) Isolation of the human BACH1 transcription regulator gene, which maps to chromosome 21q22.1. Hum Genet 102:282-288
- Busciglio J, Yankner BA (1995) Apoptosis and increased generation of reactive oxygen species in Down's syndrome neurons in vitro. Nature 378:776-779
- Cairns NJ (1999) Neuropathology of Down syndrome. J Neural Transm 57:61-74
- Chrast R, Chen H, Morris MA, Antonarakis SE (1995) Mapping of the human transcription factor GABPA (E4TF1-60) gene to chromosome 21. Genomics 28:119-122
- Chrast R, Scott HS, Chen H, Kudoh J, Rossier C, Minoshima S, Wang Y, Shimizu N, Antonarakis SE (1997) Cloning of two human homologs of the Drosophila single minded gene IM1 on chromosome 6q and SIM2 on 21q within the Down syndrome chromosomal region. Genome Res 7:615-624
- Colon EY (1972) The structure of the cerebral cortex in Down's syndrome: quantitative analysis. Neuropediatrics 3:362-376
- Crome L, Cowie V, Slater E (1966) A statistical note of cerebellar and brain stem weight in mongolism. J Ment Defic Res 10:69-77
- de Haan JB, Wolvetang EJ, lanello R, Bladier C, Kelner MJ, Kola I (1997) Reactive oxygen species and their contribution to pathology in Down syndrome. Adv Pharmacol 38:379-402
- de la Monte SM (1999) Molecular abnormalities of the brain in Down Syndrome: relevance to Alzheimer's neurodegeneration. J Neural Transm 57:1-20
- de la Monte SM, Sohn YK, Ganju N, Wands JR (1998) p53-and CD95-associated apoptosis in neurodegenerative diseases. Lab Invest 78:401-411
- Dumont A, Hehner SP, Hofmann TG, Ueffing M, Droge W, Schmitz W (1999) Hydrogen peroxide-induced apoptosis is CD-95 independent, requires the release of mitochondria-derived reactive oxygen species and the activation of NF-kappa-B Oncogene 18:747-757
- Engidawork E, Gulesserian T, Seidl R, Cairns N, Lubec G (2001) Expression of apoptosis related proteins: RAIDD, ZIP kinase, Bim/BOD, p21, Bax, Bcl-2 and NF-kB in brains of patients with Down syndrome. J Neural Transm (Suppl) 65:181-192
- Epstein CJ (1995) Down syndrome. In: Scriver CR, Beaudet AL, Sly W, Valle D (eds) The metabolic and molecular bases of inherited disease. 7th Edition, Vol. I, New York: McGraw Hill, Inc., pp 749-794
- Evan G, Littlewood T (1998) A matter of life and cell death. Science 281:1317-1322
- Gahtan E, Auerbach JM, Groner Y, Segal M (1998) Reversible impairment of long-term potentiation in transgenic Cu/Zn-SOD mice. Eur J Neurosci 10: 538-544
- Gedye A (1991) Serotonergic treatment for aggression in a Down syndrome adult showing signs of Alzheimer's disease. J Ment Defic Res 35:247-258
- Geldmacher DS, Lerner AJ, Voci JM, Noelker EA, Somple LC, Whithouse OJ (1997) Treatment of functional decline in adults with Down syndrome using selective serotonin reuptake inhibitors. J Geriatr Psychiatry Neurol 10:99-104
- Greber-Platzer S, Balcz B, Cairns N, Lubec G (1999) c-fos expression in brains of patients with Down syndrome. J Neural Transm 57:75-86
- Greber-Platzer S, Turhani-Schatzmann D, Cairns N, Balcz B, Lubec G (1999) The expression of the transcription factor ets-2 in brain of patients with Down syndrome evidence against the overexpression-gene dosage hypothesis. J Neural Transm 57:269-282
- Greber-Platzer S, Turhani-Schatzmann D, Wollenek G, Lubec G (1999) Evidence against the current hypothesis of gene dosage effects of trisomy 21. Biochem Biophys Res Comm 254:395-399
- Gulesserian T, Fountoulakis M, Seidl R, Hardmeier R, Cairns N, Lubec G (2001) Super-oxide dismutase SOD1, encoded on chromosome 21, but not SOD2 is over-expressed in brains of patients with Down syndrome. J Invest Med 49:41-46
- Gutman A, Wasylyk B, Wasylyk C (1991) Cell-specific regulation of oncogene-responsive sequences of the c-fos promoter. Mol Cell Biol 11:5381-5387
- Hansen R, Oren M (1997) p53; from inductive signal to cellular effect. Curr Opin Gene Dev, pp 46-51
- Hasegawa K, Litt L, Espanol MT, Sharp FR, Chan PH (1998) Expression of cfos and hsp70 mRNA in neonatal rat cerebrocortical slices during NMDA induced necrosis and apoptosis. Brain Res 785:262-278
- He X, Rosenfeld MG (1991) Mechanisms of complex transcriptional regulation: implications for brain development. Neuron 7:183-196
- Hockenberry DM, Oltvai ZN, Yin XM, Milliman CL, Korsmeyer SJ (1993) Bcl2 functions in an antioxidant pathway to prevent apoptosis. Cell, p. 241-251
- Ianello RC, Crack PJ, de Haan JB, Kola I (1999) Oxidative stress and neural dysfunction in Down syndrome. J Neural Transm 57:256-267
- Ivins KJ, Thornton PL, Rohn TT, Cotman CW (1999) Neuronal apoptosis induced by beta amyloid is mediated by caspase 8. Neurobiol Dis 6:440-449
- Jayanthi S, Ordonez S, McCoy MT, Cadet JL (1999) Dual mechanisms of Fas induced cell death in neuroglioma cells: a role for reactive oxygen species. Brain Mol Brain Res 72:158-165
- Jefferey KJ, Abraham WC, Dragunow M, Mason SE (1990) Induction of foslike immunoreactivity and the maintenance of long-term potentiation in the dentate gyrus of an anaesthetised rat. Brain Res Mol Brain Res 8:267-274
- Johnson TM, Yu ZX, Ferrans VJ, Lowenstein RA, Finkel T (1996) Reactive oxygen species are downstream mediators of p53-dependent apoptosis. Proc Natl Acad Sci USA 93:11848-11852
- Kaltschmidt B, Baeuerle PA, Kaltschmidt C (1993) Potential involvement of the transcription factor NF-kappa-B in neurological disorders. Mol Aspects Med 14:171-190
- Kaminska B, Mosieniak G, Gierdalski M, Kossut M, Kaczmarck L (1995) Elevated AP-1 transcription factor DNA binding activity at the onset of functional plasticity during development of rat sensory cortical areas. Brain Res Mol Brain Res 33:295-304
- Kim SH, Vlkolinsy R, Cairns N, Lubec G (2000) Decreased levels of complex III core protein 1 and complex V ß chain in brains from patients with Alzheimer's disease and Down syndrome. Cell Mol Life Sci 57:1810-1816
- Kim SH, Yoo BC, Broers JLV, Cairns N, Lubec G (2000) Neuroendocrine specific protein c, a marker of neuronal differentiation, is reduced in brain of patients with Down syndrome and Alzheimer's disease. Biochem Biophys Res Commun 276:329-334
- Kola I, Christiano F, de Haan JB, Thomas R, Sumarsono S, Corrick CM, Tymms M (1995) Genes, embryogenesis and Down syndrome. In: Moeloek FA, Affandi B, Trounson AO (eds) Advances in human reproduction. New York: Casterton-Parthenon, pp 309-320
- Labudova O, Kitzmueller E, Rink H, Cairns N, Lubec G (1999) Gene expression in fetal Down syndrome brain as revealed by subtractive hybridization. J Neural Transm 57:125-136
- Labudova O, Krapfenbauer K, Moenkemann H, Rink H, Kitzmueller E, Cairns N, Lubec G (1998) Decreased transcription factor junD in brain of patients with Down syndrome. Neurosci Lett 252:159-162
- Li PF, Dietz R, van Harsdorf R (1999) p53 regulates mitochondrial membrane potential through reactive oxygen species and induces cytochrome cindependent apoptosis blocked by Bcl-2. EMBO J 18:6027-6036
- Lubec G, Labudova O, Cairns N, Berndt P, Langen H, Fountoulakis M (1999) Reduced aldehyde dehydrogenase levels in the brain of patients with Down syndrome. J Neural Transm 57:21-40
- Lubec G (1996) The Hydroxyl Radical: From Chemistry to Disease. J Invest Med 44:324-346
- Mann DMA (1988) The pathological association between Down syndrome and Alzheimer's disease. Mech Aging Dev 43:99-136
- Mann DMA (1997) Neuropathological changes of Alzheimer's disease in persons with Down's syndrome. In: Esiri MM, Morris JM (eds) The neuropathology of dementia. Cambridge: Cambridge University Press, pp 122-136
- Marin-Padilla M (1976) Pyramidal cell abnormalities in the motor cortex of a child with Down's syndrome. J Comp Neurol 167:63-75
- Mattson MP, Partin J, Begley JG (1998) Amyloid beta induces apoptosis-related events in synapses and dendrites. Brain Res 807:167-176
- Merrill JE, Murphy SP (1997) Regulation of gene expression in the nervous system by reactive oxygen species. Metab Brain Dis 12:97-112
- Nagy Z (1999) Mechanisms of neuronal death in Down Syndrome. J Neural Transm 57:233-246
- Owen-Schaub LB, Zhang W, Cusack JC, Angelo LS, Sante SM, Fujiwara T, Roth JA, Deisseroth AB, Zhang WW, Kruzel E (1999) Wild type human p53 and a temperature sensitive mutant induce Fas/APO-1 expression. Mol Cell Biol 15:3032-340
- Peled-Kamar-M, Lotem J, Okon E, Sachs L, Groner Y (1995) Thymic abnormalities and enhanced apoptosis of thymocytes and bone marrow cells in transgenic mice overexpression Cu/Zn - superoxide dismutase: implications for Down syndrome. EMBO J 14:4985-4993
- Pennypacker KR (1995) AP-1 transcription factor complexes in CNS disorders and development. J Fla Med Assoc 82:551-554
- Pfarr CM, Mechta F, pyrou G, Lallemand D, Carillo S, Yaniv M (1994) Mouse JunD negatively regulates fibroblast growth and antagonises transformation by ras. Cell 76:747-760
- Pritchard MA, Kola I (1999) The gene dosage effect hypothesis versus the amplified developmental instability hypothesis in Down Syndrome. J Neural Transm 57:293-304
- Rehder H (1981) Pathology of trisomy 21. In: Burgio GR, Fraccaro M, Tiepolo L, Wolf U (eds) Trisomy 21. An International Symposium. Berlin. Springer Verlag, p 57
- Roberts LA, Higgins MJ, O'Shaugnessy CT, tone TW, Morris BJ (1996) Changes in hippocampal gene expression associated with the induction of long-term potentiation. Brain Res Mol Brain Res 42:123-127
- Rowen S, Fisher DE (1997) Mechanisms of apoptotic cell death. Leukemia 11:457-465
- Sawa A, Oyama F, Cairns N, Amano N, Matsushita M (1997) Aberrant expression of bcl-2 gene family in Down's syndrome brains. Brain Res Mol Brain Res 48:53-59
- Sawa A (1999) Neuronal Death in Down's syndrome. J Neural Transm 57:87-97
- Schapiro MB (1988) Dementia in Down's syndrome:cerebral glucose utilization, neuropsychological assessment and neuropathology. Neurology 38:938-942
- Schmidt-Sidor B, Wisniewski KE, Shepard TH, Sersen A (1990) Brain growth in Down syndrome subjects 15-22 weeks of gestational age and birth to 60 months. Clin Neuropathol 9:181-195
- Seidl R, Fang-Kircher S, Bidmon B, Cairns N, Lubec G (1999) Apoptosis associated proteins p53 and APO-1/Fas (CD95) in brains of adult patients with Down syndrome. Neurosci Lett 260:9-12
- Seidl R, Tiefenthaler M, Hauser E, Lubec G (2000) Effects of a single transdermal nicotine dose on cognitive performance in 5 adults with Down syndrome. Lancet 356:1409-1410
- Shapiro BL (1999) The Down syndrome critical region. J Neural Transm 57:41-60
- Sinet PM, Lejeune J, Jerome H (1979) Trisomy 21 (Down Syndrome). Glutathione peroxidase, hexose monophosphate shunt and IQ. Life Sci 24:29-39
- Sinet PM (1982) Metabolism of oxygen derivatives in Down's syndrome. Ann NY Acad Sci 396:83-94
- Strasser A, Harris AW, Cory S (1991) Bcl-2 transgene inhibits T cell death and perturbs thymic self-censorship. Cell 67:889-899
- Suzuki Y, Ono Y, Hirabayashi Y (1998) Rapid and specific reactive oxygen species generation via NADPH oxidase activation during Fas-mediated apoptosis. FEBS Lett 425:209-212
- Sylvester PE (1986) The anterior commissure in Down's syndrome. J Ment Def Res 30:19-25
- Takashima S, Ieshima A, Nakamura H, Becker LE (1989) Dendrites, dementia and the Down syndrome. Brain Dev 2:131-143
- van Leeuwen FW, Hol EM (1999) Molecular misreading of genes in Down syndrome as a model for the Alzheimer type of neurodegeneration. J Neural Transm 57:137-160
- Venugopal R, Jaiswal AK (1998) Nrf2 and Nrf1 in association with Jun proteins regulate antioxidant response element mediated expression and coordinated induction of genes encoding detoxifying enzymes. Oncogene 17:3145-3156
- Wasylyk B, Wasylyk C, Flores P, Buegue A, Leprince D, Stehelin D (1990) The cets proto-oncogene encode transcription factors that cooperate with c-Fos and c-Jun for transcriptional activation. Nature 346:191-193
- Wisniewski KE, Bobinski M (1991) Hypothalamic abnormalities in Down syndrome. In: Epstein CJ (ed) The Morphogenesis of Down syndrome. New York, Wiley-Liss, p 153
- Wisniewski KE, Laure-Kamionowska M, Connell F, Wen GY (1986) Neuronal density and synaptogenesis in the postnatal stage of brain maturation in Down syndrome. In: Epstein CJ (ed) The Neurobiology of Down syndrome. New York: Raven Press, p 29
- Wisniewski KE, Laure-Kamionowska M, Wisniewski HM (1984) Evidence of arrest of neurogenesis and synaptogenesis in Down Syndrome brains. N Engl J Med 311:1187-1188