Mini Review 2 - Biochem 5853, 1998
Oklahoma State University
|
Riverbend DS Assocation Home Page » Resources » Term Papers & Reports » Downs Syndrome Downs Syndrome |
|
Kip Karges Mini Review 2 - Biochem 5853, 1998 Oklahoma State University |
Printed with the permission of Franklin R. Leach |
Factors affecting Down syndrome metabolically
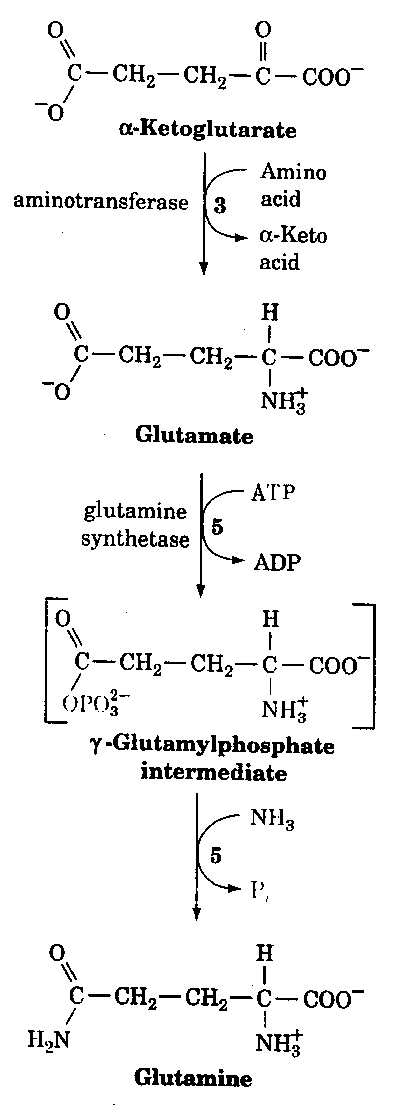
Just recently data has come out suggesting a common disease mechanism between Alzheimer's disease and Down syndrome. These mechanisms are not only through clinical and neuropathological similarities but also from a genetic and biochemical data. For example, Peeters et al.,(3) found a highly significant decrease in mitotic index in the presence of exogenous glutamine. This was observed for patients with an Alzheimer type dementia with or without an associated Down syndrome. These findings suggest that glutamine sensitivity or some dysregulation of the glutamine/glutamate pathway (Fig. 1) may play a key role in the pathogenesis of Alzheimer's disease. These findings could have important implications in the development of preventive strategies for people at risk for Alzheimer's disease.
Figure 1.
In another study (4) amyloid precursor protein (APP) content in peripheral lymphocytes was looked at to see if there is any correlation between Down syndrome (DS) and Alzheimer's disease (AD). Results indicated that patients with DS had a significantly higher, 1.5 fold increase in lymphocyte APP signal compared to normal aged-matched subjects. In contrast, both AD patients and elderly control groups had significantly increased lymphocyte APP as compared to young controls. APP immunoreactivity more than doubles from the age of 20 to 80 for non-DS subjects. This study suggest that with increased cellular APP content in DS as well as aging that it may correspond to generalized alterations in expression or processing of this molecule and possibly could be used as timing determinant of AD onset.
Researchers have also found that DS subjects are at a greater risk for developing neuropathological features of senile dementia of the Alzheimer's disease (SDAD) by middle age. Recent evidence suggest that gastoinstestinal aluminum is increased with subjects having SDAD and that aluminum may contribute to associated neuropathological changes. Although the mechanism of enhanced absorption are unknown at this time, the data indicate that similar abnormalities in the gastrointestinal handling of aluminum occurs in both SDAT and DS subjects (5).
Purines have been shown to be critical for energy metabolism, cell signaling and cell reproduction. Nevertheless, little is known about the regulation of this essential pathway during development. Purine biosynthesis is catalyzed by a trifunctional protein with glycinamide ribonucleotide synthetase (GARS), aminoimidazole ribonucleotide synthetase (AIRS) and glycinamide ribonucleotide formyltransferase (GART) enzymatic activities. The gene encoding this trifunctional protein is located on chromosome 21 (6). These results tie back into Down syndrome in that all three proteins are expressed at high levels during normal prenatal cerebellum development but become undetectable shortly after birth. In contrast, these proteins continued to be expressed during postnatal development of the cerebellum in individuals with Down syndrome. Furthermore, the premature aging seen in Down syndrome could possibly be contributed to an altered ratio between two enzymes in their organs, CU/Zn-superoxide dismutase (SOD) and glutathione peroxidase (GPX)(7). This is based on the observation that an altered Cu/Zn-SOD and GPX ratio exists in the brain of aging mice and that this correlates with increased lipid damage. Normally these enzymes serve to neutralize harmful reactive oxygen species produced during oxidative metabolism (Fig. 2) however, with an altered ratio it is postulated that this could affect the aging process in Down syndrome.
Figure 2.
Superoxide Dismutase (SOD)Conclusion
202+ 2H H2O2 + O2Glutathione peroxidase (GPX)
2GSH + R-O-O-H GSSG + ROH +H2O
References