British Osteopathic Journal 1998, Vol. XXI, 11-20
|
Nicholas J.R. Handoll, D.O. British Osteopathic Journal 1998, Vol. XXI, 11-20 |
Reprinted with the permission of the author and the British Osteopathic Journal |
This paper raises the hypothesis that postnatal hypoxia causes much of the handicap of Down's syndrome and that osteopathic treatment may be used effectively to reduce it.
It was 93 years after Landon Down in 1866 first described the syndrome which now bears his name, when Lejune in 1959 showed that this commonest form of mental handicap affecting 800 - 1,000 live births p.a. in the U.K. is due to extra genetic material carried on the long arm of chromosome 21. Down's syndrome is a congenital foetal growth disorder which affects all metabolic processes but fundamentally involves the nervous system. There are a wide range of characteristics, a small number of cases exhibit them all, some cases only a few. Some 80% of the manifestations of Down's syndrome are only minor anomalies which are commonly seen in isolation in the overall population. The effect of the extra genetic material of chromosome 21 appears to decrease the buffering to disordered development as the foetus grows, which results in a greater incidence of minor anomalies.
The fundamental deficiency in Down's syndrome involves the nervous system but the overall characteristics involve the physical appearance and behaviour as well as intellectual function. Physically, the more obvious characteristics are well known. Hypotonia, or floppiness in the newborn, is the most consistent feature. Historically, before the development of chromosomal analysis, hypotonia, the lack of normal muscle tone and response to stimuli for the age, were the most reliable first indications of the presence of Down's syndrome. The oriental expression is often not evident in the newborn.
The physical phenotype of Down's syndrome includes hypotonic muscles with flexible joints and short stature. The head is shorter in its A/P diameter, there are epicanthic folds with the characteristic slant to the eyes and a depressed nasal bridge. The lenses can be aplastic leading to refractive errors. The ears are small and the dentition is hypoplastic and immature. The neck is short. The hands have characteristic short digits, there may be clinodactyly, a simian crease and characteristic dermatoglyphic features to the palms. The feet show a gap between the first and second toes. The pelvis is hypoplastic with an outward lateral flare. The heart is anomalous in 40% of cases. The skin is dry and hair sparse. The genitalia is generally immature.
The nervous system is underdeveloped. The anterior commissure is small and immature and the dendrites of the cortical neurones are abnormal. The child suffers from learning difficulties which can vary in range in different individuals from profoundly handicapped to within normal range of intelligence.
Pueschel summarised the clinical conditions commonly observed in the Down's syndrome population from infant to adulthood. In the very young they include congenital anomalies such as cataracts, gastro-intestinal anomalies and heart disease. During early childhood common problems are infectious diseases, increased appetite, periodontitis, seizure disorders, sleep apnoea, visual impairment, audiologic deficits, thyroid dysfunction and skeletal problems. During adolescence maturation and health issues such as skin infections, thyroid disorders and increased weight gain are significant as well as mental health concerns. Similar concerns may also be observed during adulthood which is often marked also by accelerated ageing and the threat of Alzheimer's disease. [1]
Some 90% - 95% of the cases of Down's syndrome are due to trisomy of chromosome 21. This means that the individual carries a third chromosome 21 instead of the normal two
In the human there are 46 chromosomes (23 pairs) of varying size. Each chromosome consists of a pair of strands of DNA, each referred to as a chromotid. The chromotids are joined at a specific point called the centromere which is usually not at the centre, which leaves each chromotid with a short arm (known as p) and a long arm (known as q). The chromotids are joined by strands of non-genetic material known as chiasmata. Each arm of a chromosome can be divided into segments known as banding, by special staining techniques, which is used in classification. Only a small segment of chromosome 21 is responsible for the features of Down's syndrome. It is found on the long arm between 21q22.1 - 21q22.3. In other words, the small section responsible for this disorder is found between the first and third parts of the 22nd segment of the long arm of chromosome 21. This minute part of the whole human genome, which if carried as an extra segment is responsible for the features of Down's syndrome, represents perhaps only 50 - 100 genes.
There are three ways in which this disordered division can occur.
Meiotic non-disjunctionresults in the triplication of the entire 21st chromosome. During meiosis when the cells divide to produce gamete cells, if the chromotids do not segregate evenly to each pole of the cell before it divides, the daughter cells will have an abnormal number of chromosomes. This is called aneuploidy. One cell will have too few chromosomes (hypoploidy) and the other will show too many chromosomes (hyperploidy). This disorder of chromosomes is known as non-disjunction. Hypoploidy is not compatible with life. Hyperploidy is, in many cases, compatible with life and is the commonest cause of trisomy (three chromosomes rather than two).
Hyperploidy of any of the chromosomes can occur but the larger ones will have a greater effect on body metabolism so the foetus will be less able to sustain life and will be eliminated. Chromosome 21 is one of the smallest of the chromosomes so with trisomy 21 the foetus can survive. It also appears to be one of the most fragile and therefore vulnerable to translocation.
Translocation accounts for about 5% of cases and occurs when a segment of chromosomal material breaks off and attaches to another chromosome. Typically this attaches itself to one of the D group (chromosomes 13, 14 or 15), number 14 being the most common. In about 1% of cases it attaches to one of the G group, numbers 21 or 22. When the free piece has been broken from the other chromosome of the pair before separation and is therefore extra genetic material, it will cause Down's syndrome. The precise number of genes carried by the free section of chromotid can vary, but provided it contains the first, second and third parts of the 22nd segment of the long arm of 21, Down's syndrome will result.
In rare instances the broken piece of chromotid may come from the same chromosome 21 in that cell, after separation. Therefore chromosome 21 will be smaller than usual. The free piece may still attach itself to another chromosome as above, but in this case there is no trisomy and the individual is likely to be normal. This is known as balanced translocation. In even more rare cases, the free segment re-attaches to chromosome 21. Again, the individual is likely to be normal but the significance of this is in the risk of carrying the condition to the next generation. When the mother has a translocation to the D group or to chromosome 22, the risk of recurrence can be one in five births; when the father is the carrier the risk is one in twenty births. For the very rare 21/21 translocation for either parent, the risk is 100%.
Mosaicism is rare, accounting for up to 5% of Down's syndrome. It occurs when the disordered cell division takes place during early foetal development but after the first few somatic divisions. Then only a limited number of cell lines may be involved in non-disjunction or translocation so some tissues of the body will have extra genetic material and other cells will be normal. Therefore, whereas the range of abilities are likely to be similar in the vast majority of children with non-disjunction or translocation, there can be a considerable overlap in ability of children with mosaisicm.
What features of this handicap are most significant with regard to the potential for care and management? Benda, in 1960, demonstrated that the overall effect of the genetic changes in Down's syndrome is immaturity which can be explained by a slowing of growth during in the first weeks of life. Between the 6th and 12th week of interuterine life there is a critical stage of development in the processes of differentiation and development, when the growth of the Down's syndrome foetus decelerates in comparison to the norm. (fig 1) This results in a low birth weight and delay of growth and development of the brain, eyes, hands and heart. [2] Barden pointed out however that the prenatal growth retardation is small in contrast to the marked retardation characterising much of postnatal growth. [3]
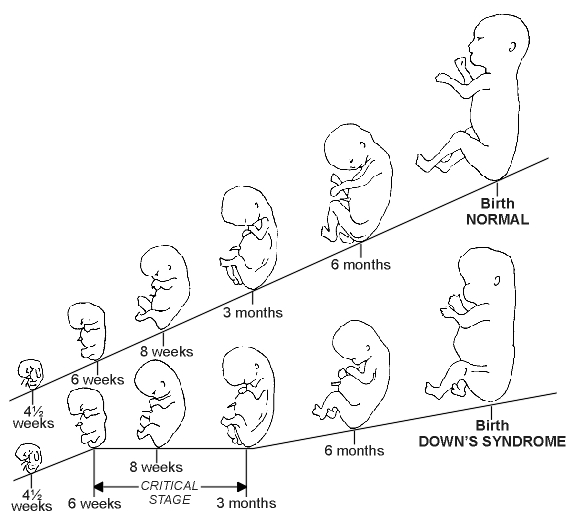 |
| Fig. 1. Growth of the Down's Syndrome Foetus Compared with Normal (redrawn after Benda [2]) Prenatal development from 4½ weeks to birth. The upper line represents normal development. The lower line represents the deceleration of normal development and differentiation seen in Down's syndrome. |
Benda showed that the initial pathology lies in the growth deficiency of the cranial base. This has been confirmed by subsequent studies. [3] This shortening of the cranial base is due to slow growth, rather than to early fusion. [3]
The development of the neurocranium and viscerocranium follow independent courses in the first few months of interuterine life but they are hinged upon the sphenoid body as the central supporting structure. The sphenoid body is the site of the tip of the embryonic notochord and the site of convergence of a number of other cranial structures, namely the midbrain and forebrain, the major transverse dural septae (the tentorium cerebelli and falx cerebri), the nasal passages and foregut. It ossifies from 14 centres through an elaborate schedule and timetable of ossification which eventually achieves the alignment of the face. This temporary interruption in the growth of the bone between the 6th and 12th weeks results in the under-development and shortening of the cranial base. It is worthwhile to note that in normal development an ossification centre appears for the lesser wings early in the 9th week and two ossification centres appear later in the 9th week for the presphenoid body. The two centres for the postsphenoid body do not appear until 16 weeks. The pre and post sphenoid bodies unite at about 32 weeks.
Factors of particular significance may be summarised as follows:
Cranial vault
At birth the Down's syndrome skull is within normal limits. Thereafter the
vault grows slowly, particularly during the first year of life. Its
circumference mirrors the pattern of normal development but always 4-5cm behind
for the age. It stops growing about the age of 14 years, when its circumference
corresponds to that of a normal child aged 3-4 years. The width of a Down's
syndrome skull is nearly normal for that age. The failure of development is
primarily due to the marked lack of growth in length. [2] (fig 2)
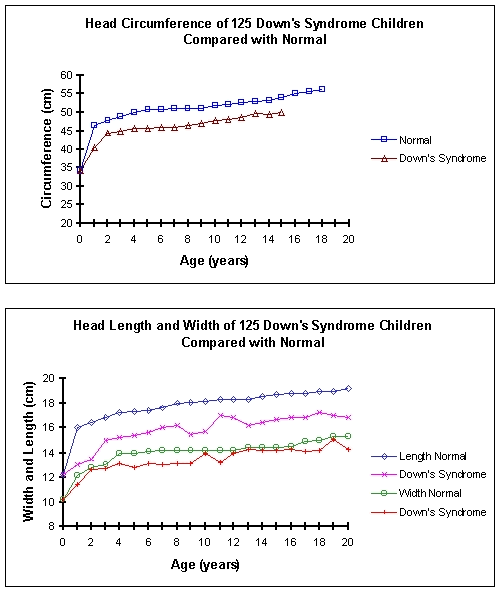 |
Head growth, indicated by mean circumference, length and width of Down's syndrome children (n=125) compared with normal, from birth to age 20 years. Circumference indicated by (normal), D (Down's syndrome), Length indicated by > (normal), X (Down's syndrome), Width indicated by O (normal), + (Down's syndrome).
|
The sutures of the vault may be separated. The saggital suture is not in approximation and the parietal bones may be separated by ½ cm. or more. A patent metopic suture is 3 times as common in cases of Down's syndrome as normal. Diploe fail to develop so that all the skull bones are thin and light. On average a Down's syndrome skull is only 3-4 mm thick, compared to a normal of 6-10 mm.
If the arrest of growth in Down's syndrome was restricted to the skull, the simplest explanation would be that the lack of development of the brain causes the lack of development of the skull. However, measurements and studies of brain development indicate that during the first half year of life the weight of the brain corresponds to normal, while concurrently there is general growth disorder of both the skull and the long bones. [2] In another study of non Down's syndrome children, no relationship was found to exist within individuals between head circumference and mental-age growth rate patterns. [4] These studies show that the volume of the vault, or neurocranium, in Down's syndrome is a little smaller than normal, but the size of the pituitary is probably within normal limits. The size of the neurocranium is not seen to be a useful indicator of mental function.
Cranial base
The body of the sphenoid is smaller than normal, particularly in its AP
dimension and the angle of the sphenoid is inclined more vertically. [5] (fig 3) The angle of the occipital squama also is steep and
upright. This abnormal angulation of the basicranium is seen in other conditions
such as Robin sequence, so is unlikely to be due entirely to a chromosomal
defect. The near vertical angle of the cranial base causes it to be displaced
forwards, which, due to the attachment to it of the pharynx, narrows the
anteroposterior dimension of the pharynx and shortens the nasal and oral
airways. This will increase airway resistance. [6, 7]
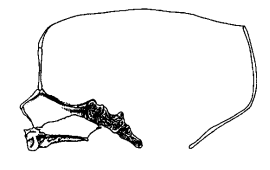 |
A. Normal foetal skull (redrawn after Bosma [4]) |
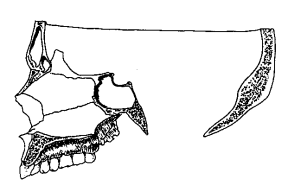 |
B. Normal adult skull (redrawn after Gray [54]) |
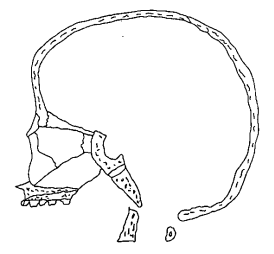 |
C. Down's Syndrome skull (redrawn after Benda [2)] |
Median Sections Through Normal and Down's Syndrome Skulls
The spheno-basilar synchondrosis and the spheno-ethmoidal articulations do not grow sufficiently and the foramen magnum is in some cases small and transversely ellipsoid. Benda showed that these changes are due to a marked lack of growth in length. There is a measurable shortening of the fronto-occipital diameter of the Down's skull, to the extent that the transverse diameter of the atlas is still in the middle of the cranial base. [2]
It may be suspected that there is a hormonal deficit. The radiographic outline of the sella turcica is not abnormal, its shape is independent of the size of the pituitary and there is no relationship of the size of the pituitary to various growth anomalies, so the lack of growth does not appear to be due to pituitary dysfunction.
Face and sinuses
All areas of the Down's syndrome skull are deficient in growth [8], but particularly in the structures which show the most marked
development after birth, i.e. the ethmoid, nasals, maxilla and mandible. The
diminutive maxillae remain retracted under the protruding forehead due to a lack
of the forward and downward thrust of growth. This delay in development results
in a persistence of foetal characteristics of the face. [2] (fig 4)
| Birth | Normal Young Adult | Down's Syndrome Age 16 |
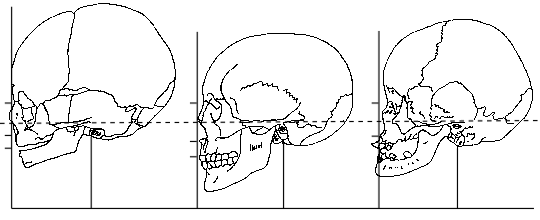 |
||
| A. | B. | C. |
Lateral Views of Normal and Down's Syndrome Skulls (redrawn after Benda [2])
In the normal individual the sinuses with the exception of the sphenoid sinus appear during the 4th-5th month of foetal life. The normal sphenoid sinus appears soon after birth in the anterior part of the body of the sphenoid and grows progressively posteriorly. The normal maxillary sinuses at birth are represented only by a furrow in the nasal wall of each bone.
In the Down's syndrome skull there is an absence in the development of the sphenoid and frontal sinuses and the maxillary antra are small. In a radiographic study of 29 Down's syndrome skulls, with an age range of 8-49 years, there was a conspicuous failure of development of the frontal sinuses in 24 cases (83%) and the sphenoid sinuses were considerably reduced in size in 66%, pneumatization being confined to the anterior part of the bone. [9] All the other sinuses, including the mastoids are also poorly formed. The cribriform plate of the ethmoid bone tends to be short and retracted, forming a small deep valley between the arches of the orbit. The absence of frontal sinuses, a particular feature of Down's syndrome, has a considerable influence on the characteristic shaping of the frontal bones and forehead.
The palate is flat inside the dental ridge but has a high dorsoventral elevation in the midline, forming what is known as a 'steeple' palate. A steeple palate is not high or flat but a combination of both. There are three important growth vectors in the development of the human palate which normally take place between the 6th - 12th week in utero. Delay in development during this period could account for these changes.
At seven weeks gestation the connective tissue floor of the embryonic nasal capsule (the early mouth, nose and cheek region) is connected to the cervical portion of the alimentary canal. Cephalically the crista galli at the base of the developing skull is anchored to the vault by the early dura mater of the falx cerebri. With the normal growth of the cerebral hemispheres during this period the head pulls off the chest, while at the same time the alimentary canal, which is resistant to tension due to its attached blood vessels, descends towards the thorax along with the hyoid region, larynx, trachea and heart. The nasal capsule, caught between these opposing forces, is stretched craniocaudally into a muzzle, like a circular rubber ring stretched vertically. This represents the first growth vector. It narrows the mouth and aligns the nostrils craniocaudally. Secondly the face deepens in an antero-posterior (dorsoventral) direction. Mesenchymal fibres in the early palatine processes each side of the tongue become stretched dorsoventrally to form straight, horizontal palatine processes. The third growth vector is the appositional growth of these paired palatine processes to form the hard palate which separates the nose from the mouth. [10]
An aberration in this timetable could disrupt the final shape of the palate, mouth and nose. Spitzer has pointed out that the high-arched palate and the shortening of nasal septum contributes to a considerable narrowing of the nasal antrum and hence a narrowed airway. [9] Because of the obstruction to the upper airway, children with Down's syndrome have to breathe through their mouth.
However, the mandible itself is small, which narrows the floor of the mouth and restricts available space. As a result of this and the flat, steeple palate, there is insufficient intra-oral space for the tongue, which therefore often protrudes. Tongue protrusion is not due to an enlargement of the tongue, which is within normal limits, but purely because of restricted space within the mouth. [9, 11] This serves to highlight how severe the upper airway obstruction in a Down's syndrome child can be.
The permanent dentition is malformed in shape and size due to the disharmonic growth of the immature dental system. The root formation is delayed with consequent delay in eruption. The teeth are stunted in growth and the crowns are shorter and smaller. They are malformed resulting in small haplodont teeth. There is aplasia of the enamel. Both the maxillary and mandibular teeth are bilaterally and equally affected. [9] In a study of 100 Down's syndrome patients it was found that 41% had missing or stunted permanent lateral incisors. [12] The shape of the tooth socket and ultimately that of the alveolar bone depends on the growth tendencies of the tooth germ. [13] It is significant to note that the maxillary laminae for the permanent lateral incisors is seen in the developing jaw at about the 9th week of interuterine life. This coincides with the critical stage at which the processes of differentiation and development in the Down's syndrome foetus decelerates.
The high incidence of oral and dental anomalies, all of which are seen in isolated incidences in the general population, shows how the trisomy has the effect of, not so much creating the anomaly per se, but of decreasing the developmental and physiological buffering against genetic and environmental forces. In other words the chromosomal imbalance has a negative effect on the stability of developmental pathways. [3, 14, 15, 16]
About half the cases of Down's syndrome will suffer from conductive deafness and otitis media which may be associated with anatomical abnormalities in the middle ear. About 20% of cases will be sensorineural and this incidence increases with age. The overall length of the cochlea is shorter in Down's syndrome and the spiral ganglion cell population is reduced. In the middle ear there may be found abnormalities such as maldevelopment of the stapes and facial canal dehiscence. [17]
It has been stated already that the fundamental disorder of Down's syndrome involves the nervous system. The evidence suggests that there is no difference from normal in the density of cortical neurones in the full term Down's syndrome infant but the dendritic spines on individual neurones degenerate from the early post-natal period onwards. [18] This is a premature arrest of growth so that by the time the individual reaches adulthood, the deficiency in dendritic spines is severe. A loss such as this appears to have a significant effect on neurobehavioural retardation whether in cases of Down's syndrome or not. In a study of cortical biopsies from 5 young children with severe neurobehavioural retardation of unknown aetiology, it was found that the normal cylindrical geometry in individual neurone dendritic processes was interrupted by varicosities. This was accompanied by thin and irregular proximal processes, loss of dendritic spines and the predominance of long, thin tortuous spines. It was concluded that alternations in dendritic structure of cortical neurones may represent a primary target in the pathobiological process underlying neurobehavioural failure. [19] In a parallel computer reconstruction study it was found that the microtubules become disorientated. They may form into helical swirls and become discontinuous in the varicose regions. [20]
It has been noted that most pyramidal neurones in the hippocampus should have acquired a full complement of spines by 6 months of age. Severe metabolic and cardiorespiratory disturbances and/or chromosomal abnormalities can influence significantly dendritic spine morphology and development. [18] Purpura studied the stratum pyramidale in the hippocampus of a 1.5 week old infant with Down's syndrome and Tetralogy of Fallot. Apart from a reduction of neurone cell size there was no difference in the density of neurones, but there was a striking absence of the axonal plexus. His conclusions were that the decreased density or loss of the axonal plexus of the stratum pyramidale is probably not due directly to the genetic abnormality of Down's syndrome. One of a number of possible contributing factors was the congenital cyanotic heart disease, which could conceivably influence the perinatal development of the axonal plexus. In a 6-week old full term normal chromosomal infant with prolonged and unremitting cyanotic heart disease the axonal plexus was virtually absent despite the presence of well-developed pyramidal neurones. He concluded that prolonged cerebral hypoxaemia, with associated blood and tissue pH changes, is a significant obstruction to the postnatal development of synaptic pathways. [18]
Purpura makes the following observations:
In such conditions as Down's syndrome cortical neuronal growth and differentiation including early synaptogenesis may proceed with some temporal delays through the last phases of antenatal development and perhaps through most of the first year of infancy. Thereafter degenerative changes in elements of the neuropil (axonal terminals, axodendritic and axospinodendritic synapses) may occur at a variable tempo with consequent loss of or bizarre alterations in dendritic spines, the appearance of dendritic branching pattern abnormalities, and attenuation and degeneration of axonal terminals.[18]
It seems unlikely that specific abnormalities in dendritic spine structure characterize the dendritic spines in cases of specific chromosomal abnormalities known to be associated with mental retardation. The same may be said in regard to the development of the axon plexus of the hippocampus. Prematurity and associated cardiac anomalies and/or prolonged neonatal hypoxemia may be more important in influencing the development of the axon plexus than the presence of Down's syndrome per se.[18]
What is being suggested here is that the neurone density in the Down's syndrome neonate cortex appears to be close to normal limits, but the subsequent degeneration of the neurone dendrites and axon plexus, which is associated with neurobehavioural failure, appears to be largely a result of postnatal hypoxaemia. There are a number of clinical studies which reflect the same conclusion.
Several authors have shown that upper airway resistance or obstructive sleep apnoea in children is likely to result in hypoxaemia, both in children with Down's syndrome [21, 22] or without Down's syndrome. [23, 24, 25] This is exacerbated by upper respiratory tract infections. [24]
Southall found that six out of twelve children with Down's syndrome had sleep related upper airway obstruction and five of these had episodes of hypoxaemia even while breathing continuously. Four of the six had measurably reduced 'background' levels of blood oxygen saturation during regular breathing, compared to controls. [26]
Several authors have shown that obstructive sleep apnoea in children with hypoxaemia results in a wide range of behavioural disturbances and neurodevelopmental problems such as hyperactivity, aggression, learning difficulties, school failure, neurodevelopmental delay, daytime somnolence, failure to thrive and even permanent neurological damage. The extra effort and energy expenditure required to breathe results in tiredness and fatigue in adults and behavioural disturbances and failure to thrive in children. [7, 27, 28] A narrowed airway is a common feature of Down's syndrome [Stebbens VA, Personal communication.][9], so many of these problems are common in Down's children. [21, 22] If left untreated they can result in a "slow death". [22, 26] However, similar problems can also occur in children who do not have Down's syndrome if the airway is compromised. [25, 30, 31, 32, 33, 34, 35] In cases of Robin sequence it has been noted that failure to thrive is almost always related to upper airway obstruction. [7, 27] This lends further support to the proposal that it is not the chromosomal anomaly directly which causes the behavioural disturbances and neurodevelopmental delay but the obstructive sleep apnoea and hypoxaemia. It is a question therefore whether the mental handicap associated with Down's syndrome is a direct consequence of the trisomy alone or whether breathing difficulties in early childhood are in some part to blame. Loughlin questions whether the growth failure and developmental delay associated with inherited craniofacial anomalies may not be a secondary phenomenon attributable to obstructive sleep apnoea. [30]
The conventional treatment in cases of obstructive sleep apnoea is adenotonsillectomy to open the airway. [31] In some cases this achieves remarkable results [33, 36, 37] and in some cases a growth spurt is reported. [25, 32, 33] However in a proportion of children adenotonsillectomy is not entirely satisfactory [21, 22, 38, 39, 40] and more extensive surgery such as tracheostomy [26, 38, 41], uvulopalatopharyngoplasty [22, 30, 42, 43], reduction of the tongue [30, 35] or maxillofacial or maxillo-mandibular surgery [30, 35] has been used. These cases can present postoperative complications. [35, 44] Stebbens has said that there is a need for better treatment in severe cases. [21] More conservative measures, such as continuous positive airway pressure, have been tried [37, 45] but this is difficult to administer and can present technical and compliance complications. [28, 35, 38, 46]
Even cardiac disease in Down's children cannot be put down entirely to congenital causes. [33, 47] Guilleminault and Suzuki observed haemodynamic changes during obstructive apneas in nocturnal sleep, which they considered caused 24-hour hypertension. [48] They found that tracheostomy eliminated the hypertension in these children. Marcus et al. concluded that hypoxaemia in some children with Down's syndrome may result in pulmonary hypertension and congestive heart failure. She found that many Down's children have pulmonary hypertension unassociated with, or out of proportion to, congenital heart disease. [22] Chi and Krovetz came to the same conclusion. [49] Perkin and Anas describe how chronic alveolar hypoventilation from upper airway obstruction can result in pulmonary arterial hypertension, and right ventricular hypertrophy. A vasoconstrictive response to the chemical changes from hypoxaemia and hypercapnia as a result of hypoventilation induces hypertrophy of the muscles in the pulmonary vessels. The increase in the resistance increases the work of the right ventricle, which eventually develops right ventricular hypertrophy and heart failure. Hypoxic vasoconstriction is more profound in the infant than in the adult. Once pulmonary hypertension and cor pulmonale are established the condition is self perpetuating, but in its early stages it is reversible. [50, 33] Heart disease due to hypoxaemia can mimic congenital heart disease. Levine & Simpser cite four Down's syndrome infants who developed cor pulmonale and heart failure in association with upper airway obstruction, which masqueraded as cyanotic congenital heart disease. [41] Tracheostomy provided a marked benefit to three of the four. Other studies report similar findings in infants [7, 30, 33, 34, 35, 36, 47] and in adults. [51]
Down's syndrome is a congenital foetal growth disorder due to trisomy 21, affecting all metabolic processes. Its effects are an immaturity of growth and decreased physiological buffering against disordered development from environmental forces.
The nervous system is affected primarily but research has shown that most of the degeneration takes place after birth. Neurone degeneration is associated with hypoxaemia due to a combination of narrowed airways reducing pulmonary respiration and heart disease impairing circulation. One may speculate that the Down's syndrome foetus is protected to some degree by the maternal respiratory system. Research suggests that reduced oxygen uptake is highly significant and may precipitate neurological damage and pulmonary heart disease, even in otherwise normal infants. It is reasonable to conclude that the provision of a constant adequate airway from the moment of birth with the minimum of invasion is highly desirable as a first line of treatment for an infant with Down's syndrome.
There is a clear sequence here. It is unlikely that this sequence alone is the cause of the complete pattern of defects found in Down's syndrome but it is probable that it has some part to play. The sequence begins with the delay in development at 6-12 weeks in utero. The full term infant is therefore immature, particularly with regard to the nervous system and the cranial base, but essentially the tissues at this moment are largely sound. However, the delay in the growth and development of the cranial base continues, resulting in insufficient growth of the maxillary bones and the upper airway. The facial sinuses are also under developed, particularly the sphenoid, frontal and maxillary sinuses. This results in poor drainage of mucus, which congeals, can become infected and causes further difficulty in breathing. The further narrowing of the airway leads to sleep apnoea and hypoxaemia. Chronic hypoxaemia appears to be a major cause of maldevelopment of the cortical neurone dendrites and axonal bed. This is associated with neuro-developmental delay, and such behavioural problems as poor learning, hyperactivity and aggression. Children with such behaviour require special schooling. The prospect is that they will become classified as educationally subnormal and require special care throughout their life.
There is a window in this sequence which may be accessed by an appropriately skilled osteopath. The window is open immediately after birth.
In 1939 William Sutherland, an osteopathic physician from Kirksville, Missouri, USA, proposed the concept of the primary respiratory mechanism in which he postulated that the bones of the cranium were flexible and allowed some movement against each other. [52, 53] It is known that live, wet bone has elastic, compressive and tensile properties which will allow for deformation. [54] In 1987, Retzlaff & Mitchell showed that intracranial sutures contain connective tissue, Sharpey's fibres, blood vessels and nerves. [55] Sutherland's hypothesis allows for only tiny amounts of 'give' between individual cranial bones, much of the shape change being taken up by the deformation of the bone itself, but that is all the change required. This phenomenon includes the bones of the face.
It is known that the epithelial lining of the facial sinuses contain goblet cells which secrete mucus and ciliated cells which waft the mucus away into the nasopharynx. Sutherland postulated that in addition each sinus has one or more other bones which pump the sinus clear mechanically rather like a suction plunger used to clear a blocked sink drain. Clinical experience shows that when this mechanism is compromised by mechanical or other forces the sinus is less able to drain its mucus, which pools, thickens and becomes a hotbed for infection. The nasal mucosa becomes continually inflamed with a copious purulent discharge, [42] and the adenoids and tonsils enlarge. Frequently antibiotics are prescribed which reduce the infection and discharge but do nothing for the stasis of mucus within the sinus. In the Down's syndrome child, the narrowed airway is further narrowed or even blocked by the discharge, forcing the child to breath through its mouth already narrowed by the steeple palate, shortened by the steep angle of the cranial base and the micrognathia It is perhaps not surprising that, as a result, the youngster's tongue protrudes.
Mechanical dysfunction such as this can be detected by the palpating fingers of a competent osteopath adequately trained in Sutherland's development of osteopathic medicine. The practitioner can release the dysfunction by gentle, conservative and non-invasive means. Personal clinical experience has shown that once the mechanical dysfunction has been released and the physiological mechanical pumping of the affected sinuses restored, the sinuses are able to drain. Subsequently the hyperaemic mucous membrane shrinks and the swollen adenoids and tonsils return to normal. The frequency of attacks of sinusitis reduces and with it the need for repeated prescriptions of antibiotics, because there is no stasis of mucus to become infected. The airway is clear. There are no contra-indications to treatment, administered by a properly trained osteopath, with regard to the age of the patient.
A 7 year 5 month old girl with Down's syndrome was brought to see whether anything could be done to aid her poor general health. She had eaten very little during the first four years of her life but since then her appetite had improved. She presented as a sickly child, of low intelligence, with a characteristic blocked nose.
After one year and 7 treatments her father reported that "she is better than she has ever been". After just over two years from consultation and a further 10 treatments her father described her as "thriving". She was attending a normal primary school for three days a week and a special school for the remaining two days. After a further three treatments she was discharged with instructions for her to return whenever her breathing became compromised, particularly at night.
Eighteen months later, aged nearly 12 years, her father reported that her health was "robust". She had received no further osteopathic treatment but her breathing was still clear. She had suffered no respiratory infections, she had taken no days away from school, nor even seen her doctor for two years. She was still educationally subnormal for her age but she could talk clearly and fluently in 4-5 word sentences and joined in conversations. She was still attending the same schools and was popular with the other children. She was an accepted member of her local Girl Guides unit. She lives with her family and her parents are delighted with her progress.
A 5 year 4 month old boy with Down's syndrome presented with constant rhinorrhea, enlarged adenoids, sleep apnoea and bilateral otitis media. An atrioventricular septal defect, diagnosed soon after birth, was repaired successfully. This was followed by a bilateral cleft lip repair and a cleft palate repair. At one year of age he was found to have bilateral narrow external auditory meati and middle ear effusions which were treated at the age of two years with bilateral grommets. The otitis media continued however and at the time of consultation he was on a waiting list for a third pair of grommets. He had required inhalers periodically for chest infections. Nocturnal pulse oximetry at the age of 5 showed that his overall oxygen saturation remained good throughout the night but there were several brief dips to 75% saturation. These periods were self rectifying as he would turn over and start breathing again. It was decided that surgery was not indicated.
He had 5 osteopathic treatments over a period of four months. Each time his breathing improved with short periods of a clear airway and good night's sleep. During the next four months (February to May) he suffered recurrent upper respiratory tract infections and attended only three sessions. There was a 90 minute journey each way and the boy's parents discontinued treatment.
Osteopathic treatment will not stop acute rhinitis, but will tend to increase the discharge and help clear the infected material and hence shorten the attack. Improvement in the chronic condition will continue once the acute infection has resolved. His was a promising start bearing in mind his medical history. It was to be expected that the periods of clear breathing would become steadily longer and more frequent. However in this case treatment was not administered for sufficient time to break the chronic discharge / infection cycle. A year and a half later, at the age of 7 years and 6 months, his adenoids are of normal size but he still has a discharge from his nose.
The patient was a girl with Down's syndrome who is now 18 years old. She was brought in for treatment first as a two week old baby. She was already breathing through her mouth and her tongue was protruding. She was treated at intervals throughout her baby and toddler years to encourage her body to drain her facial sinuses and clear her nose. By the time she was 6 years old her airway was clear. At the age of 7 years she was assessed for sleep apnoea and found to be normal.
She attended a Waldorf school until she was 16. She enjoyed her time there and was popular with the other children. However she was found to be dyslexic, as are both her father and her otherwise normal elder sister, and she found it difficult to learn to read and write to the extent that her dyslexia gave her far more difficulties at school than the Down's syndrome. A special needs teacher was appointed for her and she did not attempt GCSE examinations.
At the age of 16 she gained a place at a specialist boarding school for youngsters with special needs of all types, including physical disabilities, which is geared towards training for independent life skills and employment. She was the first in her year to be moved out of the school buildings into independent accommodation with four other youngsters, where they are responsible for their own daily personal routine. She can do her own washing and ironing and can prepare her own breakfast and simple snacks. Soon she will be buying her own simple groceries.
Her mother says that she is more reliable and mature in her life skills and self responsibility, although her fellow students can communicate with the written word more effectively than she. Her general health is excellent and she does not contract the colds and chest problems to which her peers are prone. She did not develop any heart defects. She had stopped having regular osteopathic treatment by the time she was 9 years old, after which her mother would bring her for treatment whenever she had an upper respiratory tract infection or before an airline flight. One pre-flight treatment would prevent her from suffering from in-flight earache. The day after treatment for an upper respiratory tract infection her nose would stream, "like turning the taps on" said her mother. The next day the infection would clear. Her last bad cold was two years ago, which resolved after one treatment.
Now aged 18 she does not have skin problems as she has no nasal discharge and her mother believes that she does not have the characteristic facial features of Down's syndrome. Her daughter, she says, "has a proper nose". (fig 5)
 |
 |
| Age 4 Months | Age 17 Years |
It has been shown that hypoxaemia, particularly during the first few years of life, has a bearing on the subsequent degeneration of the dendrites and axonal plexus of the neurones of the central nervous system and that this contributes to the development of neurodevelopmental delay, neurobehavioural difficulties or even premature death. It has also been shown that one of the effects of the decreased buffering against disordered development from environmental forces due to trisomy 21 is a delay in the development of the face, particularly the maxillary bones which retain foetal like proportions, and the mal-development or non-development of the major facial sinuses. Clinical experience suggests that this is associated with an increased mechanical tension of the somatic tissues of the face and neurocranium which is not found in the normal population. This is thought to involve the anterior pole of the falx cerebri. This effect is known to osteopaths as a compression.
It is speculated that chromosome induced reduction in buffering leading to mal-development of the face and facial sinuses may lead, either completely or in part, to the mechanical effects of the compression involving the falx cerebri and somatic tissues of the face. An osteopath trained in this field should be able to prevent or reduce the mal-development of the face and facial sinuses in the Down's syndrome neonate. Provided appropriate treatment is commenced soon after birth, this should allow fluids and mucus to drain effectively from the facial sinuses, preventing infection, blockage, epithelial hyperaemia and enlarged adenoids and tonsils. Better oxygenation should take place, to allow the cortical neurones to develop. This simple measure alone could reduce the neurodevelopmental delay so common in the Down's syndrome child. In the longer run the child may be able to attend normal schools, develop normally within the community and possibly eventually become an independent human being able to fend for him or herself in the community, even able to earn his or her own living.
It is anticipated that osteopathic treatment should be administered as appropriate for the first 18 months of life while the sinuses are beginning to form and thereafter whenever the sinuses become congested, until puberty. Against that should be put the saving in the parents' time caring for the child, the cost of special schooling, the cost to the health service in medication, surgery and other specialist services, the cost to the unemployment services and the cost of care during the lifetime of the individual. It is speculated even, that by reducing the compression in the region of the sphenoid and frontal sinuses, appropriate treatment may have the effect of reducing the depression at the bridge of the nose, perhaps the dominant facial deviation of the human being with trisomy 21.
This paper raises the hypothesis that postnatal hypoxia causes much of the handicap of Down's syndrome and that osteopathic treatment may be used effectively to reduce it. Evidence is presented to support the proposal that much of the handicap of Down's syndrome is not due directly to the chromosomal defect, but to impaired postnatal development as a result of hypoxaemia from upper airway obstruction. It is proposed that appropriate treatment from birth given by suitably trained osteopaths can offer a safe, non-invasive and effective means of maintaining a patent airway. It is postulated that this will reduce some of the severe and widespread disabilities which handicap the individual with trisomy 21.
I should like to thank Simon J. Dunmore, PhD, Senior Lecturer in Biomedical Sciences, School of Health Sciences, University of Wolverhampton, for his invaluable critique of this paper.
Nicholas Handoll, D.O.
Holland House,
70, Belmont Road,
Hereford, HR2 7JW,
England.
E-mail: handoll@o-s-l.com