Cornelius Ani* MBBS MSc DCH, Research Fellow;
Sally Grantham-McGregor MBBS MD, Professor of Child Health and Nutrition, Centre for International Child Health;
David Muller PhD, Reader in Biochemistry, Biochemistry Unit, Institute of Child Health, University College London, London, UK.
|
 |
Reprinted with the permission of Michael Pountney
Developmental Medicine & Child Neurology March 2000, 42(3): 207-213
Copyright © 2000 Mac Keith Press
|
*Correspondence to first author at Centre for International Child Health, Institute of Child Health, University College London, 30 Guilford Street, London WC1N IEH, UK.
E-mail: cani@ich.ucl.ac.uk
Down syndrome (DS) is the most common inherited cause
of learning disability1, and affected individuals are more
prone to infections, leukaemia, congenital heart disease and
other anomalies, thyroid dysfunction, early senescence and
Alzheimer's disease2. These complications create a heavy
burden for carers of individuals with DS and the social and
health services. Consequently, any intervention that ameliorates
some of these complications will have a significant
impact on the quality of life of individuals with DS and their
carers. However, before investing resources in interventions,
it is important to ascertain their scientific validity.
The Internet and the lay press make many claims that certain
expensive nutritional supplements improve the outcome
in DS. These claims have left some health professionals
confused and parents of children with DS vulnerable to pressures
to spend large amounts of money on nutritional supplements
whose benefits have not been proved. This review
was undertaken firstly, to determine whether there is a theoretical
basis to expect that nutritional supplements may
improve the pathology of DS, and secondly, to critically
examine published trials of nutritional supplements in DS to
determine whether the existing evidence supports claims
that nutritional supplements improve the outcome in DS.
Theoretical basis for supplementation
OXIDATIVE STRESS IN DS
Oxidative stress is defined as an imbalance between production
of oxygen-derived free radicals and their removal by
antioxidants3. The activity of superoxide dismutase (SOD) —
a key enzyme in the metabolism of oxygen-derived free radicals
— is increased in DS (see below). This increase in SOD
activity may alter the normal steady state equilibrium of reactive
oxygen species leading to oxidative injury in DS4,5.
SOD catalyses the dismutation of superoxide radicals to
hydrogen peroxide which is further metabolised to water by
glutathione peroxidase (GSH-Px) or catalase3. In DS, the
SOD:GSH-Px ratio is increased and this imbalance may lead
to the accumulation of unmetabolised hydrogen peroxide
which may then react with transition metals like iron (Fenton
reaction)3 to form the hydroxyl radical6 (Fig. 1). The latter is
the most reactive oxygen radical known and can, for example,
readily initiate lipid peroxidation resulting in damage to
cell membranes3. Because the brain is rich in highly polyunsaturated
fatty acids which are particularly susceptible to
lipid peroxidation, it is potentially vulnerable to this type of
damage7. Therefore, it is hypothesised that an overexpression
of SOD causes free radical mediated damage and that
this may contribute to the learning disability and early onset
of Alzheimer's disease which are typical of DS. The following,
which will be considered in turn, support the above hypothesis:
(1) there is an increase in SOD activity, (2) there is evidence
of increased lipid peroxidation, (3) there is a
compensatory increase in the activities of GSH-Px and the
hexose monophosphate shunt (HMPS).
Increased superoxide dismutase (SOD) activity
The presence of an extra chromosome 21 in DS results in
overexpression of genes residing on that chromosome. The
resulting 'gene dose effect' is thought to account for most of
the pathophysiology of DS1. One of these genes codes for the
enzyme SOD 6 . As expected from the gene dose effect, many
studies have found an increase in SOD activity of 50% or more in various tissues of individuals with DS8-16.
Similarly, many animal studies have shown 50% or more
overexpression of SOD in transgenic mouse models of
DS5,17-20.
Increased lipid peroxidation
A number of animal studies have shown that an increase in
SOD is associated with increased rates of lipid peroxidation
in the brain5,17,21-23.
A 36% increase in lipid peroxidation was also demonstrated
in the cerebral cortex of fetuses with DS who were incubated
in vitro with iron and ascorbate compared with infants
without DS12. In addition, Busciglio and Yankner24 showed
that cultured cortical neurons from fetuses with DS had
approximately four times more intracellular free radicals and
an increased level of lipid peroxidation compared with neurons
from individuals without DS. The neurons of fetuses
with DS were also more likely to undergo apoptotic degeneration
which was prevented by the addition of antioxidants.
Significantly higher levels of the products of lipid peroxidation
have also been reported in the blood or urine of individuals
with DS than in individuals without DS13,25-27.
Compensatory increase in the activity of glutathione
peroxidase and the hexose monophosphate shunt
In addition to increased SOD activity, many studies have
reported increased activity of GSH-Px in various tissues of
individuals with DS6,7,11,13,1628-33, as well as in animal
models18. Because Wijnen and colleagues34 have localised
the gene for GSH-Px on chromosome 3, its increase in individuals
with DS is not a gene dose effect. Therefore, it is likely
to be a physiological/protective response to cope with the
excess hydrogen peroxide produced by the hyperactive SOD
system6. However, the 50% increase in activity of SOD is much
higher than the percentage increase usually reported in activity
of GSH-Px6,7,16,32,33. De Haan and colleagues35 reported a
two-fold elevation in the SOD:GSH-Px ratio measured in a
number of organs of aborted fetuses with DS compared with
control fetuses. These authors also showed that cells with a
high SOD:GSH-Px ratio are more likely to undergo apoptosis
when challenged with hydrogen peroxide, which supports
the findings of Busciglio and Yankner24 cited earlier.
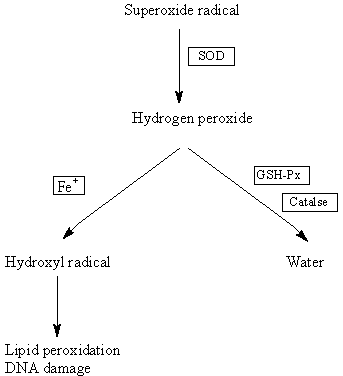
Figure 1: Hypothesised pathway for increase in SOD
leading to increased oxidative stress in DS.
Further evidence of increased oxidative stress in DS was
provided by Sinet and coworkers7 who demonstrated a 15%
increase in HMPS activity in the red blood cells of individuals
with DS compared with individuals without DS. Because
HMPS is involved in the metabolic pathway that sustains the
activity of GSH-Px, its increased activity in individuals with
DS is further evidence for the presence of oxidative stress7.
An increase in HMPS activity has also been reported in mice
transgenic for SOD23.
OXIDATIVE STRESS AND OTHER FEATURES OF DS
Immune dysfunction
It has been hypothesised that abnormal metabolism of reactive
oxygen species may also contribute to the defective
immunity and increased susceptibility to infections typically
seen in individuals with DS. The formation of oxygen radicals
is one of the key mechanisms by which phagocytic leukocytes
kill pathogens36. The serious consequence of failure in
this system is clearly seen in the dramatic increase in severe
infections of patients with chronic granulomatous disease37 whose leukocytes cannot form the superoxide radical
because of a deficiency of nicotinamide adenine dinucleotide
phosphate oxidase38.
There are at least two possible mechanisms by which an
increase in SOD activity can reduce immunity in DS. Firstly, a
hyperactive SOD system is likely to result in a decrease in the
concentration of superoxide radicals, which may in turn
cause a reduction in the microbicidal activity of leukocytes23,36. Secondly, an increase in SOD activity may lead to
an excess of hydrogen peroxide which may damage immune
cells and impair normal signal transduction processes
involved in phagocyte activation23.
In support of these hypotheses, it has been shown that
neutrophils from individuals with DS produce less superoxide
radicals than individuals without DS36,39. Similarly,
Mirochnitchenko and Inouye 23 found that a two-fold over-production
of SOD by intraperitoneal macrophages from
transgenic mice, resulted in an inhibition of extracellular
release of superoxide radicals, increased intracellular production
of hydrogen peroxide, and a reduction in microbicidal
and fungicidal activity.
Peled-Kamar and colleagues5 have shown that the activity
of SOD in the thymus of transgenic mice is increased by two-to
five-fold, and that the thymus is more susceptible to
lipopolysaccharide-induced apoptotic cell death. The
increased SOD activity was also associated with an increased
production of hydrogen peroxide and lipid peroxidation.
When cultured under stressed conditions (e.g. addition of
tumour necrosis factor), the bone marrow cells from the
transgenic mice produced two to three times fewer granulocyte
and macrophage colonies than control mice. It was suggested
that these defects resulted from increased oxidative
damage5 although the authors did not investigate whether
the addition of antioxidants corrected the immune defects.
Malignancy
There is now evidence linking increased oxidative stress with
increased DNA damage in DS27,40. Jovanovic and coworkers27 compared the levels of 8-hydroxy-2-deoxyguanosine (a biomarker
of oxidative damage to DNA) in 166 matched pairs of
individuals with DS and their siblings, and found a significantly
increased concentration of 8-hydroxy-2-deoxyguanosine in
the urine of individuals with DS. Pincheira and colleagues40
found an increase in chromosomal damage in lymphocytes of
individuals with DS compared with individuals without DS,
which could be reduced by more than 50% by the addition of
vitamin E to the cell culture. As vitamin E is a powerful antioxidant,
it was hypothesised that the increased chromosomal
damage in DS resulted from increased oxidative stress. These
studies, therefore, not only provide further direct evidence
for increased oxidative stress in DS but also suggest a possible
explanation for the increased malignant potential associated
with the syndrome.
Mental development
There is some evidence of an association between mental
development in DS and oxidative stress. Sinet and
colleagues7 found a highly significant positive correlation
between GSH-Px activity and IQ in 22 individuals with DS and
concluded that GSH-Px may play an important role in preserving
the cerebral status of individuals with DS. As GSH-Px
is an endogenous antioxidant, it is possible that supplementing
individuals with DS with exogenous antioxidants may
offer similar protection to their cerebral status. This is supported
by the protective effect of antioxidants on DS neurons
in culture, referred to earlier24. A randomised controlled trial
of vitamin E in Alzheimer's disease41 found significant beneficial
effects. Because individuals with DS almost invariably
develop Alzheimer's disease, this trial suggests a role for
oxidative damage in the pathology of both conditions.
Premature ageing
De Haan and colleagues35 have carried out several studies
suggesting that increased oxidative stress could be implicated
in ageing. They found (1) a significant increase in the
SOD:GSH-Px ratio (p<0.005) and susceptibility to lipid peroxidation
(p>0.005) in normal mouse brain during ageing,
(2) that cultured murine cells which have been transfected to
have an increased SOD:GSH-Px ratio showed the characteristic
features of senescence, (3) that normal mouse cells
exposed to hydrogen peroxide in culture also showed features
of senescence, and (4) that cells derived from children
with DS showed features of senescence which were not seen
in cells from age-matched control children.
CONCLUSIONS AND SOME IMPLICATIONS OF INCREASED OXIDATIVE STRESS IN DS
In summary, the evidence for increased oxidative stress in DS
is reasonably strong and includes: gene dose overexpression
of SOD, increased lipid peroxidation in human individuals
with DS and transgenic mice models, compensatory increases
in GSH-Px and HMPS activities, increased products of
oxidative DNA damage, increased chromosomal damage
reduced by 50% in vitro by the addition of vitamin E, and
most importantly there are increased intracellular free radicals
and enhanced apoptosis in neurons of fetuses with DS
which can be prevented by addition of antioxidants.
Therefore, there is good evidence that increased oxidative
stress may play a role in the complications of DS. This means
that an excess of oxygen-derived free radicals could result in
an extra demand for antioxidant nutrients like vitamins C
and E, ß-carotene, and selenium (cofactor for GSH-Px). Thus
even normal serum concentrations of these nutrients could
be functionally deficient in the face of excess demand. This
opens the possibility that antioxidant nutrient supplementation
might help to ameliorate the pathology of DS. We
would, therefore, hypothesise that supplementation with
increased amounts of antioxidant nutrients could benefit
individuals with DS. The following section reviews trials of
nutritional supplementation and a selection of other non-nutritional/pharmacological interventions which have been
carried out in individuals with DS.
Nutritional supplementation trials in DS
We found many published trials of supplementation with
nutrients and pharmacological agents in individuals with DS,
including zinc, selenium, megavitamin/mineral preparations,
vitamin A, vitamin B6 and its precursors, 'targeted
nutritional intervention (TNI) supplements', vasopressin,
and 'U series' (see below). The results have been varied and
will now be briefly reviewed. Unfortunately, only a few of the
studies were randomised trials and although, as indicated
above, there is a theoretical rationale for antioxidant supplementation,
none of the trials was specifically designed to
evaluate antioxidant therapy.
ZINC SUPPLEMENTATION
Zinc is part of the cytosolic copper-zinc-SOD enzyme6. Zinc
supplementation trials have been justified mainly because of
reports of relatively low serum zinc in DS. Of 16 studies
which have compared serum levels of zinc in individuals with
DS and individuals without DS, 13 showed significantly
reduced zinc concentrations in individuals with DS42-54, while three found no significant difference55-57.
We found only one randomised controlled trial of zinc in
DS58. These investigators assigned 64 individuals with DS
aged 1 to 19 years to receive 25 to 50 mg of zinc/day (depending
on age) or placebo for 6 months with a crossover for
another 6 months. Outcome criteria consisted of laboratory
measures of immune competence and an infection log which
included respiratory symptoms such as coughing. They
found no significant changes in lymphocyte function, complement
levels, or number of infections. However, the trend
was in favour of the zinc-treated group (p=0.07) for days
coughed and they had significantly (p=0.03) fewer episodes
of cough. Also, among children less than 10 years old, the
zinc-treated group had significantly fewer cough days
(p=0.01) than placebo controls.
There have been seven uncontrolled zinc trials with pre-and
posttreatment measurements with a total of 168 individuals
with DS aged 2 to 22 years43,45,47,49-51,53. All the studies
consistently reported mainly laboratory evidence for beneficial
effects of zinc supplementation on the immune function
of individuals with DS. However, as these studies had no
placebo treated controls or blind assessment of outcome,
they are difficult to interpret.
We found two in vitro studies with zinc in DS. Fabris and
colleagues44 reported that adding zinc to the serum of individuals
with DS increased their serum thymic factor (FTS) to
levels normally seen in individuals without DS and also
reduced the concentration of FTS inhibitory factor. Chiricolo
and coworkers59 showed that individuals with DS who were supplemented for 4 months with 1 mg of zinc/kg/day had an
increase in the in vitro incorporation of thymidine into their
phytohaemagglutinin (PHA) stimulated lymphocytes similar
to individuals without DS. In addition, following gamma
radiation induced damage to DS cells, zinc supplementation
reduced the abnormally high DNA repair rate (which predisposes
to mutations and increases malignant potential) in DS
cells to normal levels59. However, as these studies were conducted
in vitro their in vivo significance remains uncertain.
In summary, although there are encouraging results from
uncontrolled studies and in vitro experiments suggesting
that zinc supplementation may enhance immunity and
reduce malignant potential in individuals with DS, there is
no rigorous or consistent evidence from clinical trials to
show that this is the case.
SELENIUM SUPPLEMENTATION
Selenium is a component of GSH-Px which is part of the
body's endogenous antioxidant system6. In a study by
Annerén and coworkers60, 10 mg of selenium/kg/day was
administered to 48 individuals with DS aged 1 to 16 years for
6 months, and concentrations of immunoglobulin G2 and
G4 increased by up to 33% and 75% respectively. The participants
also reported fewer infections during the study.
However, as this study was uncontrolled and almost half of
the sample was lost during follow-up, the result is impossible
to interpret. In another study, Antila and coworkers61 gave 15
to 25 µg of selenium/kg/day to seven individuals with DS
aged 1 to 54 years for a period of 0.3 to 1.5 years and
reported a 25% increase in the activity of GSH-Px and a 24%
reduction in the SOD:GSH-Px ratio compared with 10
unsupplemented individuals with DS.
MEGAVITAMIN/MINERAL SUPPLEMENTATION
In 1981, Harrell and colleagues62 randomised 22 children
aged 5 to 15 years with learning disability (five of whom had
DS) to receive either a megavitamin/mineral preparation or
placebo for 4 months initially. After the first phase, all the participants
received the megavitamin/mineral supplement for
another 4 months. The supplement consisted of 11 vitamins
and eight minerals in high doses and included two antioxidants:
vitamin C, 1500 mg; and vitamin E, 600 IU, daily. The
investigators reported dramatic improvements in IQ,
growth, physical appearance, language, educational attainment,
and general health of the treated participants. This
study had significant problems in that the loss of participants
reduced the already small sample from 22 to 16 and only four
of these had DS. However, the findings stimulated several
more trials of megavitamin/mineral supplementation.
Six randomised controlled trials63-68 attempted to replicate
the findings of Harrell and coworkers62 using similar vitamin/
mineral supplements. These studies consisted of a
total of 161 individuals with DS aged between 6 months and
40 years and none of the studies showed any improvement in
IQ, physical appearance, or general health.
VITAMIN A SUPPLEMENTATION
We found only one small trial of vitamin A (retinol) in DS.
Palmer69 paired 23 individuals with DS aged 3 to 15 years with
their own siblings and randomly assigned each pair to receive
either 1000 IU/kg/day of vitamin A or placebo for 6 months. At
baseline, the participants with DS in both groups experienced significantly more frequent infections than their siblings
(p<0.01). However, during follow-up, the difference in
frequency of infections between the vitamin A treated participants
with DS and their siblings gradually reduced, becoming
insignificant (p>0.05) by the fifth month of the study. In contrast,
the difference remained significant (p<0.01) between
the untreated participants with DS and their siblings throughout
the study. However, the analyses were difficult to interpret
because the treated and control groups were not
statistically compared and the frequency of infections of individual
children were summed. Also, it was not clear if the
assessment of infections was performed 'blind'. This trial was
conducted on the basis of reports of poor absorption and
reduced serum vitamin A concentration in DS25,56,69.
However, this rationale is weak as impaired absorption of vitamin
A was not reported in a larger study 70 and others have
not found reduced serum vitamin A concentrations14,70-73.
VITAMIN B6 / 5-HYDROXYTRYPTAMINE (5-HTP) SUPPLEMENTATION
Individuals with DS have been treated with vitamin B6 or 5-
HTP in order to increase their serotonin level74, which is frequently
reported to be reduced75-77. Despite two
uncontrolled studies76,78 which reported improvements in
the muscle tone of 23 babies and children with DS treated
with 5-HTP, two randomised controlled trials74,79 failed to
find any significant clinical improvements in a total of 108
babies with DS treated with vitamin B6 or 5-HTP for 3 years.
TARGETED NUTRITIONAL INTERVENTION (TNI) SUPPLEMENTATION
Supplementation with TNI is probably the most popular nutritional
therapy currently advocated for individuals with DS
judging by its extensive coverage on the Internet and in lay
publications. Its proponents claim to have identified the biochemical
abnormalities in DS and have formulated a supplement
to 'target' these abnormalities. A typical TNI supplement
contains about 56 nutrients including vitamins, minerals,
enzymes, amino acids, electrolytes etc. Unfortunately, we
found no published trial on the safety or efficacy of this supplement.
In addition, we found that a typical TNI preparation
contains 1000 mg of vitamin C which may be unsafe in children,
given that a daily intake of 500 mg of vitamin C has been
shown to have pro-oxidant effects in adults80.
MISCELLANEOUS TREATMENT TRIALS IN DS
In addition to the nutritional supplements discussed above,
individuals with DS have also been treated with various pharmacological
agents and two of these will now be briefly
reviewed.
Following reports that vasopressin enhances learning in
animals81, Eisenberg and colleagues82 treated nine individuals
with DS, aged 10 to 42 years, with vasopressin or placebo
for 10 days using a double-blind randomised crossover
design. They found no significant improvements in tests of
learning or memory.
Bumbalo and coworkers83 conducted a double-blind randomised
controlled trial of a preparation called the
'U series of drugs' on 24 children with DS aged 3 months to 11 years
and reported no significant treatment effects after 1 year. The
'U series of drugs' was developed by Henry Turkel84 and has
been a popular therapy for DS in many countries. The supplement
contained 48 items which, in addition to vitamins and
minerals, included substances such as rutin, naphazoline hydrochloride, propyl paraben, and pentylene tetrazole. No
theoretical rationale was given for most of the items included
in this supplement.
Comment on published supplementation trials in DS
Almost all the supplementation studies discussed above
had major methodological shortcomings. In most studies,
the design was poor and only a few were randomised controlled
trials. Many lacked control subjects, had small sample
sizes, ran for too short a duration, and targeted older
individuals with DS.
None of the studies had a sample size large enough to
detect small treatment effects, and thus they were all prone
to type II statistical error85. We have calculated the minimum
sample size required to detect a 6-point (half a standard deviation)
difference in IQ to be 170 individuals with DS (i.e. 85
in each of the treatment and control groups), assuming a
power of 90% and 5% level of significance.
Most of the studies were of short duration. It may be too
optimistic to expect subtle physiological improvements to be
translated into detectable physical and mental changes within
a short time period. For example, in a clinical trial of
vitamin E in Alzheimer's disease an interim analysis performed
after 1 year showed no significant treatment effects but significant
effects were subsequently observed after 2 years41.
In most of the trials reviewed, the study participants had a
very wide age range and comprised of older children and
adults. Scientific pragmatism would suggest that the best
outcomes would be among the youngest participants in
whom the brain is developing rapidly and before damage has
been done by the complications of DS. Wisniewski and colleagues86 have shown that pathological changes in the brain
of children with DS begin in late pregnancy which suggests
that interventions to limit this damage should begin soon
after birth. Thus most previous investigators may have studied
individuals who have been too old to benefit maximally.
As already noted, some of the studies lacked a sound theoretical
basis so that they had no scientific rationale to expect
treatment effects and even if observed, there would have
been no rational or plausible scientific explanation.
Conclusion
There is an increasingly good body of evidence to suggest
that increased oxidative stress may be involved in the pathology
of DS. Therefore, it is theoretically possible that using
antioxidant nutrients to scavenge oxygen-derived free radicals
may ameliorate some of the complications of DS.
Despite this possibility there have been no clinical trials
which have specifically evaluated the effects of antioxidant
nutrient supplementation on the health and development of
children with DS. Indeed, we believe that to date there has
been no consistent or rigorous proof that any form of nutritional
supplementation improves the outcome in DS. There
is, therefore, an urgent need for a well conducted clinical
trial to evaluate the hypothesis that antioxidant supplementation
may improve the outcome in DS.
Accepted for publication 20th January 2000.
Acknowledgements
CA was part funded by the Down's Syndrome Research Foundation
and SGM is part funded by the Department for International
Development.
References
- Epstein CJ. (1995) Down syndrome (trisomy 21). In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The Metabolic and Molecular Bases of Inherited Disease Volume 1. New York: McGraw Hill. p 749-94.
- Marder E, Dennis J. (1997) Medical management of children with Down's syndrome. Current Paediatrics 7: 1-7.
- Halliwell B, Gutteridge JMC. (1989) Free Radicals in Biology and Medicine. 2nd edn. Oxford: Clarendon Press.
- Kedziora J, Bartosz G. (1988) Down's syndrome: a pathology involving the lack of balance of reactive oxygen species. Free Radical Biology and Medicine 4: 317-30.
- Peled-Kamar M, Lotem J, Okon E, Sachs L, Groner Y. (1995) Thymic abnormalities and enhanced apoptosis of thymocytes and bone marrow cells in transgenic mice overexpressing Cu/Zn-superoxide dismutase: implications for Down syndrome. EMBO Journal 14: 4985-93.
- Sinet PM. (1982) Metabolism of oxygen derivatives in Downs syndrome. In: Sinex FM, Merril CR, editors. Alzheimer's Disease, Downs Syndrome and Ageing. New York: The New York
Academy of Sciences. p 83-94.
- Sinet PM, Lejeune J, Jerome H. (1979) Trisomy 21 (Down's
syndrome). Glutathione peroxidase, hexose monophoshate
shunt and IQ. Life Sciences 24: 29-33.
- Sinet PM, Lavelle F, Michelson AM, Jerome H. (1975) Superoxide
dismutase activities of blood platelets in trisomy 21. Biochemical
and Biophysical Research Communications 67: 904-9.
- Crosti N, Serra A, Rigo A, Viglino P. (1976) Dosage effect of SOD-A
gene in 21-trisomic cells. Human Genetics 31: 197-202.
- Feaster WW, Kwok KW, Epstein CJ. (1977) Dosage effects for superoxide dismutase-1 in nucleated cells anueploid for
chromosome 21. American Journal of Human Genetics
29: 563-70.
- Bjorksten B, Marklund S, Hagglof B. (1984) Enzymes of
leukocyte oxidative metabolism in Down's syndrome. Acta
Paediatrica Scandinavica 73: 97-101.
- Brooksbank BW, Balazs R. (1984) Superoxide dismutase, glutathione peroxidase and lipid peroxidation in Down's syndrome fetal brain. Brain Research 318: 37-44.
- Kedziora J, Bartosz G, Gromadzinska J, Sklodowska M,
Wesowicz W, Scianowski J. (1986) Lipid peroxides in blood
plasma and enzymatic antioxidative defence of erythrocytes in
Down's syndrome. Clinica Chimica Acta 154: 191-4.
- Tanabe T, Kawamura N, Morinobu T, Murata T, Tamai H, Mino M,
Takai T. (1994) Antioxidant enzymes and vitamins in Down's
syndrome. Pathophysiology 1: 93-7.
- De-la-Torre R, Casado A, Lopez-Fernandez E, Carrascosa D,
Ramirez V, Saez J. (1996) Overexpression of copper-zinc
superoxide dismutase in trisomy 21. Experientia 52: 871-3.
- Pastor MC, Sierra C, Dolade M, Navarro E, Brandi N, Cabre E,
Mira A, Seres A. (1998) Antioxidant enzymes and fatty acid status
in erythrocytes of Down's syndrome patients. Clinical
Chemistry 44: 924-9.
- Ceballos-Picot I, Nicole A, Briand P, Grimber G, Delacourte A,
Defossez A, Javoy-Agid F, Lafon M, Blouin J, Sinet PM. (1991)
Neuronal-specific expression of human copper-zinc superoxide
dismutase gene in transgenic mice: animal model of gene
dosage effects in Down's syndrome. Brain Research
552: 198-214.
- Ceballos I, Delabar JM, Nicole A, Lynch RE, Hallewell RA,
Kamoun P, Sinet PM. (1988) Expression of transfected human
CuZn superoxide dismutase gene in mouse L cells and NS20Y
neuroblastoma cells induces enhancement of glutathione
peroxidase activity. Biochimica et Biophysica Acta Gene
Structure and Expression 949: 58-64.
- Barkats M, Bertholet JY, Venault P, Ceballos-Picot I, Nicole A,
Phillips J, Moutier R, Roubertoux P, Sinet PM, Cohen-Salmon C.
(1993) Hippocampal mossy fiber changes in mice transgenic for
the human copper-zinc superoxide dismutase gene.
Neuroscience Letters 160: 24-8.
- Nabarra B, Casanova M, Paris D, Nicole A, Toyama K, Sinet PM,
Ceballos I, London J. (1996) Transgenic mice overexpressing
the human Cu/Zn-SOD gene: ultrastructural studies of a
premature thymic involution model of Down's syndrome
(trisomy 21). Laboratory Investigation 74: 617-26.
- Elroy-Stein O, Bernstein Y, Groner Y. (1986) Overproduction of
human Cu/Zn-superoxide dismutase in transfected cells:
extenuation of paraquat-mediated cytotoxicity and
enhancement of lipid peroxidation. EMBO Journal 5: 615-22.
- Ceballos-Picot I, Nicole A, Clement M, Bourre JM, Sinet PM.
(1992) Age-related changes in antioxidant enzymes and lipid
peroxidation in brains of control and transgenic mice
overexpressing copper-zinc superoxide dismutase. Mutation
Research 275: 281-93.
- Mirochnitchenko O, Inouye M. (1996) Effect of overexpression
of human Cu Zn superoxide dismutase in transgenic mice on macrophage functions. Journal of Immunology 156: 1578-86.
- Busciglio J, Yankner BA. (1995) Apoptosis and increased generation of reactive oxygen species in Down's syndrome
neurons in vitro. Nature 378: 776-9.
- Shah SN, Johnson RC, Singh VN. (1989) Antioxidant vitamin (A
and E) status of Down's syndrome subjects. Nutrition Research
9: 709-15.
- Bras A, Monteiro C, Rueff J. (1989) Oxidative stress in trisomy
21. A possible role in cataractogenesis. Ophthalmic Paediatrics
Genetics 10: 271-7.
- Jovanovic SV, Clements D, MacLeod K. (1998) Biomarkers of
oxidative stress are significantly elevated in Down syndrome.
Free Radical Biology and Medicine 25: 1044-8.
- Sinet PM, Michelson AM, Bazin A, Lejeune J, Jerome H. (1975b)
Increase in glutathione peroxidase activity in erythrocytes from
trisomy 21 subjects. Biochemical and Biophysical Research
Communications 67: 910-15.
- Agar NS, Hingston J. (1980) Glutathione peroxidase activity in
red blood cells from subjects with Down's syndrome and non-mongoloid
mental retardation. Medical Journal of Australia
1: 556-7.
- Kedziora J, Lukaszewicz R, Koter M, Bartosz G, Pawlowska B,
Aitkin D. (1982) Red blood cell glutathione peroxidase in simple
trisomy 21 and translocation 21/22. Experientia 38: 543-4.
- Neve J, Sinet PM, Molle L, Nicole A. (1983) Selenium, zinc and
copper in Down's syndrome (trisomy 21): blood levels and
relations with glutathione peroxidase and superoxide
dismutase. Clinica Chimica Acta 133: 209-14
- Neve J, Vertongen F, Cauchie P, Gnat D, Molle L. (1984)
Selenium and glutathione peroxidase in plasma and
erythrocytes of Down's syndrome (Trisomy 21) patients.
Journal of Mental Deficiency Research 28: 261-8.
- Gromadzinska J, Wasowicz W, Sklodowska M. (1988) Glutathione peroxidase activity, lipid peroxides and selenium status in blood in patients with Down's syndrome. Journal of Clinical Chemistry and Clinical Biochemistry 26: 255-8.
- Wijnen LMM, Monteba-Van Heuvel M, Pearson PL, Meera Khan P.
(1978) Assignment of a gene for glutathione peroxidase (GPX1) to
human chromosome 3. Cytogenetics and Cell Genetics 22: 232-5.
- de Haan JB, Wolvetang EJ, Iannello R, Bladier C, Keiner MJ, Kola I.
(1997) Reactive oxygen species and their contribution to
pathology in Downs syndrome. Advances in Pharmacology
38: 379-402.
- Annerén G, Bjorksten B. (1984) Low superoxide levels in blood
phagocytic cells in Down's syndrome. Acta Paediatrica
Scandinavica 73: 345-8.
- Mills EL, Quie-PG. (1980) Congenital disorders of the function
of polymorphonuclear neutrophils. Reviews of Infectious
Diseases 2: 505-17.
- Baehner RL. (1996) Chronic granulomatous disease. In:
Behrman RE, Kliegman RM, Arvin AM, editors. Nelson Textbook
of Pediatrics. Philadelphia: WB Saunders Company. p 596-8.
- Kedziora J, Blaszczyk J, Sibinska E, Bartosz G. (1990) Down's
syndrome: increased enzymatic antioxidative defence is
accompanied by decreased superoxide anion generation in
blood. Hereditas 113: 73-5.
- Pincheira J, Navarrete MH, de la Torre C, Santos MJ. (1999)
Effect of vitamin E on chromosomal aberrations in lymphocytes
from patients with Downs syndrome. Clinical Genetics
55: 192-7.
- Sano M, Ernesto C, Thomas RG, Klauber MR, Schaffer K,
Grundman M, Woodbury P, Growdon J, Cotman CW, Pfeiffer E, et
al. (1997) A controlled trial of selegiline, alpha-tocopherol, or
both as treatment for Alzheimer's disease. New England Journal
of Medicine 336: 1216-22.
- Milunsky A, Hackley BM, Halsted JA. (1970) Plasma, erythrocyte
and leucocyte zinc levels in Down's syndrome. Journal of
Mental Deficiency Research 14: 99-105.
- Bjorksten B, Back O, Gustavson KH, Hallmans G, Hagglof B,
Tarnvik A. (1980) Zinc and immune function in Down's
syndrome. Acta Paediatrica Scandinavica 69: 183-7.
- Fabris N, Amadio L, Licastro F, Mocchegiani E, Zannotti M,
Francheschi C. (1984) Thymic hormone deficiency in normal
ageing and Down's syndrome: is there a primary failure of the
thymus? Lancet 1: 983-6.
- Franceschi C, Chiricolo M, Licastro F, Zannotti M, Masi M,
Mocchegiani E, Fabris N. (1988) Oral zinc supplementation in
Down's syndrome: restoration of thymic endocrine activity and
of some immune defects. Journal of Mental Deficiency Research
32: 169-81.
- Purice M, Maximilian C, Dumitriu I, Ioan D. (1988) Zinc and
copper in plasma and erythrocytes of Down's syndrome
children. Endocrinologie 26: 113-17.
- Stabile A, Pesaresi MA, Stabile AM, Pastore M, Sopo SM, Ricci R,
Celestini E, Segni G. (1991) Immunodeficiency and plasma zinc
levels in children with Down's syndrome: a long-term follow-up
of oral zinc supplementation. Clinical Immunology and
Immunopathology 58: 207-16.
- Rascon Trincado MV, Lorente Toledano F, Salazar A-Villalobos V.
(1992) Evaluation of plasma zinc levels in patients with Down
syndrome. Anales Espanoles De Pediatria 37: 391-3.
- Licastro F, Mocchegiani E, Zannotti M, Arena G, Masi M, Fabris N.
(1992) Zinc affects the metabolism of thyroid hormones in
children with Down's syndrome: normalization of thyroid
stimulating hormone and of reversal triiodothyronine plasmic
levels by dietary zinc supplementation. International Journal of
Neuroscience 65: 259-68.
- Licastro F, Mocchegiani E, Masi M, Fabris N. (1993)
Modulation of the neuroendocrine system and immune
functions by zinc supplementation in children with Down's
syndrome. Journal of Trace Element and Electrolytes in
Health and Disease 7: 237-9.
- Licastro F, Chiricolo M, Mocchegiani E, Fabris N, Zannoti M,
Beltrandi E, Mancini R, Parente R, Arena G, Masi M. (1994) Oral
zinc supplementation in Down's syndrome subjects decreased
infections and normalized some humoral and cellular immune
parameters. Journal of Intellectual Disability Research
38: 149-62.
- Sustrova M, Strbak V. (1994) Thyroid function and plasma immunoglobulins in subjects with Down's syndrome (DS)
during ontogenesis and zinc therapy. Journal of
Endocrinological Investigation 17: 385-90.
- Brigino EN, Good RA, Koutsonikolis A, Day NK, Kornfeld SJ.
(1996) Normalization of cellular zinc levels in patients with
Down's syndrome does not always correct low thymulin levels.
Acta Paediatrica 85: 1370-2.
- Kadrabova J, Madaric A, Sustrova M, Ginter E. (1996) Changed serum trace element profile in Down's syndrome. Biological
Trace Element Research 4: 201-6.
- McBean LD, Smith JC, Berne BH, Halsted JA. (1974) Serum zinc
and alpha2-macroglobulin concentration in myocardial
infarction, decubitus ulcer, multiple myeloma, prostatic
carcinoma, Down's syndrome and nephrotic syndrome. Clinica
Chimica Acta 50: 43-51.
- Matin MA, Sylvester PE, Edwards D, Dickerson JWT. (1981)
Vitamin and zinc status in Down's syndrome. Journal of Mental
Deficiency Research 25: 121-6.
- Yarom R, Sherman Y, Sagher U, Peled IJ, Wexler MR, Gorodetsky
R. (1987) Elevated concentrations of elements and
abnormalities of neuromuscular junctions in tongue muscles
of Down's syndrome. Journal of the Neurological Sciences
79: 315-26.
- Lockitch G, Puterman M, Godolphin W, Sheps S, Tingle AJ,
Quigley G. (1989) Infection and immunity in Down's syndrome:
a trial of long-term low oral doses of zinc. Journal of Pediatrics
114: 781-7.
- Chiricolo M, Musa AR, Monti D, Zannotti M, Franceschi C.
(1993) Enhanced DNA repair in lymphocytes of Down's
syndrome patients: the influence of zinc nutritional
supplementation. Mutation Research 295: 105-11.
- Annerén G, Magnusson CG, Nordvall SL. (1990) Increase in serum concentrations of IgG2 and IgG4 by selenium
supplementation in children with Down's syndrome. Archives
of Disease in Childhood 65: 1353-5.
- Antila E, Nordberg U, Syväoja E, Westermarck T. (1990)
Selenium therapy in Down Syndrome: a theory and a clinical
trial. Advances in Experimental Medicine and Biology
264: 183-6.
- Harrell RF, Capp RH, Davis DR, Peerless J, Ravitz LR. (1981) Can nutritional supplements help mentally retarded children? An exploratory study. Proceedings of the National Academy of Sciences USA. 78: 574-8.
- Bennett FC, McClelland S, Kriegsmann EA, Andrus LB, Sells CJ.
(1983) Vitamin and mineral supplementation in Down's syndrome. Pediatrics 72: 707-13.
- Coburn SP, Schaltenbrand WE, Mahuren DJ, Clausman RJ,
Townsend D. (1983) Effect of megavitamin treatment on mental
performance and plasma vitamin B6 concentrations in mentally
retarded young adults. American Journal of Clinical Nutrition
38: 352-5.
- Ellis NR, Tomporowski PD. (1983) Vitamin/mineral
supplements and intelligence of institutionalized mentally
retarded adults. American Journal of Mental Deficiency
88: 211-14.
- Smith GF, Spiker D, Cicchetti D, Justice P. (1983) Failure of vitamin/mineral supplementation in Down's syndrome. Lancet 2: 41. (Letter.)
- Weathers C. (1983) Effects of nutritional supplementation on IQ and certain other variables associated with Down's syndrome. American Journal of Mental Deficiency 88: 214-17.
- Bidder RT, Gray P, Newcombe RG, Evans BK, Hughes M. (1989)
The effects of multivitamins and minerals on children with Down's syndrome. Developmental Medicine & Child Neurology
31: 532-7.
- Palmer S. (1978) Influence of vitamin A nutriture on the
immune response: findings in children with Down's syndrome.
International Journal of Vitamin and Nutrition Research
48: 188-216.
- Pueschel SM, Hillemeier C, Caldwell M, Senft K, Mevs C,
Pezzullo JC. (1990) Vitamin A gastrointestinal absorption in
persons with Down's syndrome. Journal of Mental Deficiency
Research 34: 269-75.
- Barden HS. (1977) Vitamin A and carotene values of
institutionalized mentally retarded subjects with and without
Down's syndrome. Journal of Mental Deficiency Research
21: 63-74.
- Storm W. (1990) Hypercarotenaemia in children with Down's
syndrome. Journal of Mental Deficiency Research 34: 283-6.
- Cutress TW, Mickleson KN, Brown RH. (1976) Vitamin A
absorption and periodontal disease in trisomy G. Journal of
Mental Deficiency Research 20: 17-23.
- Coleman M, Sobel S, Bhagavan HN, Coursin D, Marquardt A, Guay M, Hunt C. (1985) A double blind study of vitamin B6 in
Down's syndrome infants. Part 1 - Clinical and biochemical
results. Journal of Mental Deficiency Research 29: 233-40.
- Tu J, Zellweger H. (1965) Blood-serotonin deficiency in Down's
syndrome. Lancet 2: 715-17.
- Bazelon M, Paine RS, Cowie VA, Hunt P, Houck JC, Mahanand
D. (1967) Reversal of hypotonia in infants with Down's
syndrome by administration of 5-hydroxytryptophan. Lancet
1: 1130-3.
- Godridge H, Reynolds GP, Czudek C, Calcutt NA, Benton M.
(1987) Alzheimer-like neurotransmitter deficits in adult Down's
syndrome brain tissue. Journal of Neurology, Neurosurgery and
Psychiatry 50: 775-8.
- Petre-Quadens O, De Lee C. (1975) 5-Hydroxytryptophan and
sleep in Down's syndrome. Journal of the Neurological Sciences
26: 443-53.
- Pueschel SM, Reed RB, Cronk CE, Goldstein BI. (1980)
5-hydroxytryptophan and pyridoxine. The effects in young
children with Down's syndrome. American Journal of Diseases
of Children 134: 838-44.
- Podmore ID, Griffiths HR, Herbert KE, Mistry N, Mistry P, Lunec J.
(1998) Vitamin C exhibits pro-oxidant properties. Nature 392: 559.
(Letter.)
- Rigter H, Crabbe JC. (1979) Modulation of memory by pituitary
hormones and related peptides. Vitamins and Hormones
37: 153-241.
- Eisenberg J, Hamburger-Bar R, Belmaker RH. (1984) The effect
of vasopressin treatment on learning in Down's syndrome.
Journal of Neural Transmission 60: 143-7.
- Bumbalo TS, Morelewicz HV, Berens DL, Buffalo MD. (1964)
Treatment of Down's syndrome with the "U" series of drugs.
Journal of American Medical Association 187: 361.
- Turkel H, Nusbaum I. (1985) Medical Treatment of Down Syndrome and Genetic Diseases. 4th edn. Southfield, Michigan: Ubiotica.
- Kirkwood BR. (1988) Essentials of Medical Statistics. London:
Blackwell Science.
- Wisniewski KE, Kida E, Ted Brown W. (1996) Consequences of
genetic abnormalities in Down's syndrome on brain structure
and function. In: Rondal JA, Perera J, Nadel L, Comblain A,
editors. Down's Syndrome. Psychological, Psychobiological, and Socio-educational Perspectives. London: Whurr Publishers Ltd. p 3-19.